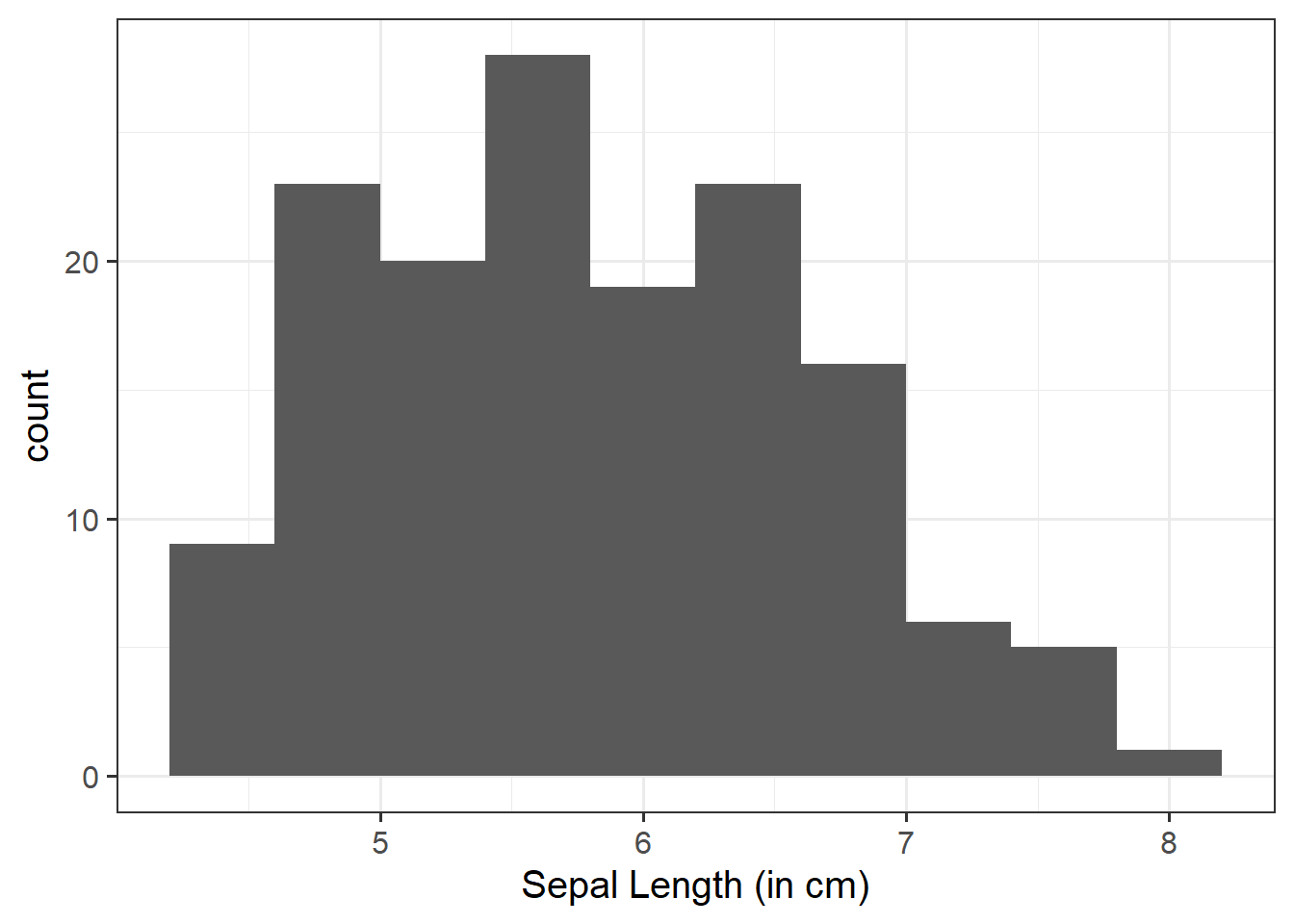
Within this reading, the following packages are used:
tidyverse
sjPlot
kableExtra
psych
patchwork
plotly
Presenting Results
Note that you must not copy any of the write-ups included below for future reports - if you do, you will be committing plagiarism, and this type of academic misconduct is taken very seriously by the University. You can find out more here.
Back to Basics
For an overview of basic statistical tests and core concepts (e.g., \(p\)-values), please revisit the DAPR1 materials for a refresher (also accessible via the DAPR1 Learn page).
Terminology
Let’s spend some time to remind ourselves of some key terminology, specifically related to types of variables and study designs:
Term
Definition
(Observational) unit
The individual entities on which data are collected
Variable
Any characteristic recorded on the observational units
Numeric variable
A variable that records a numerical quantity for each case. For such variables standard arithmetic operations make sense. For example: height, IQ, and weight
Categorical variable
A categorical variable places units into one of several groups. For example: country of birth, dominant hand, and eye colour
Binary variable
A special case of categorical variable with only 2 possible levels. For example: handedness (left or right), smoking status (smoker or non-smoker), pass test (yes or no)
Response variable (also more commonly called a dependent variable, or outcome variable)
Measures the outcome of interest in a study
Explanatory/independent variable (also called predictors)
Are used to explain differences/changes in the response variable
Observational study
An observational study is a study in which the researcher does not manipulate any of the variables involved in the study, but merely records the values as they naturally exist
Experimental study
An experiment is a study in which the researcher imposes the values of the explanatory variable on the units before measuring the response variable
Data Exploration
The common first port of call for almost any statistical analysis is to explore the data, and we can do this visually and/or numerically.
Marginal Distributions & Bivariate Associations
Marginal Distributions
Bivariate Associations
Description
The distribution of each variable individually (i.e., without reference to the values of the other variables).
Describing the association between two numeric variables.
The shape of the distribution. Look at the shape, centre and spread of the distribution. Is it symmetric or skewed? Is it unimodal or bimodal?
Identify any unusual observations. Do you notice any extreme observations (i.e., outliers)?
Plot associations among two variables.
You could use, for example, geom_point() for a scatterplot to comment on and/or examine:
The direction of the association indicates whether there is a positive or negative association
The form of association refers to whether the relationship between the variables can be summarized well with a straight line or some more complicated pattern
The strength of association entails how closely the points fall to a recognizable pattern such as a line
Unusual observations that do not fit the pattern of the rest of the observations and which are worth examining in more detail
Numerically
Compute and report summary statistics e.g., mean, standard deviation, median, min, max, etc.
You could, for example, calculate summary statistics such as the mean (mean()) and standard deviation (sd()), etc. within summarize()
Numeric exploration of data involves examining and describing key statistics like mean, median, and standard deviation via descriptives tables; and assessing the associations among variables through correlation coefficients. Exploring our data numerically helps us to identify patterns and associations in the data. When doing so, it is important to contextualise the descriptive statistics within the scope of the research question and associated scales.
Descriptives
Descriptives Tables
There are numerous packages available that allow us to pull out descriptive statistics from our dataset such as tidyverse and psych.
When we pull out descriptive statistics, it is useful to present these in a well formatted table for your reader. There are lots of different ways of doing this, but one of the most common (and straightforward!) is to use the kable() function from the package kableExtra.
This allows us to give our table a clear caption (via caption = "insert caption here", align values within columns e.g., center aligned via align = "??"), and we can also round to however many decimal places we desire (standard for APA is 2 dp; via digits = ??).
We can also add in the function kable_styling(). This is really helpful for customsing your table e.g., the font size, position, and whether or not you want the table full width (as well as lots of other things - check out the helper function!).
We can use the summarise() function to numerically summarise/describe our data. Some key values we may want to consider extracting are (though not limited to): the mean (via mean(), standard deviation (via sd()), minimum value (via min()), maximum value (via max()), standard error (via se()), and skewness (via skew()).
library(tidyverse)library(kableExtra)# using the pre-loaded iris dataset# taking the mean and standard deviation of sepal length via the summarize function# returning a table with a caption, where numbers are rounded to 2 dp# asking for a table that is not the full width of the window displayiris|>summarize( M_Length =mean(Sepal.Length), SD_Length =sd(Sepal.Length))|>kable(caption ="Sepal Length Descriptives (in cm)", digits =2)|>kable_styling(full_width =FALSE)
Sepal Length Descriptives (in cm)
M_Length
SD_Length
5.84
0.83
library(tidyverse)library(kableExtra)# using the pre-loaded iris dataset# grouping by Species. NOTE: we can group by 2 variables - we would just separate by a comma within group_by( , )# taking the mean and standard deviation of sepal length via the summarize function# returning a table of sepal length grouped by species with a caption, where numbers are rounded to 2 dp# asking for a table that is not the full width of the window displayiris|>group_by(Species)|>summarize( M_Length =mean(Sepal.Length), SD_Length =sd(Sepal.Length))|>kable(caption ="Sepal Length (in cm) Grouped by Species Descriptives Table", digits =2)|>kable_styling(full_width =FALSE)
Sepal Length (in cm) Grouped by Species Descriptives Table
Species
M_Length
SD_Length
setosa
5.01
0.35
versicolor
5.94
0.52
virginica
6.59
0.64
The describe() function will produce a table of descriptive statistics. If you would like only a subset of this output (e.g., mean, sd), you can use select() after calling describe() e.g., describe() |> select(mean, sd).
library(psych)library(kableExtra)# using the pre-loaded iris dataset# we want to get descriptive statistics of the iris dataset, specifically the sepal length column# we specifically want to select the mean and standard deviation from the descriptive statistics available (try this without including this argument to see what values you all get out)# returning a table with a caption, where numbers are rounded to 2 dp# asking for a table that is not the full width of the window displaydescribe(iris$Sepal.Length)|>select(mean, sd)|>kable(caption ="Sepal Length Descriptives (in cm)", digits =2)|>kable_styling(full_width =FALSE)
Sepal Length Descriptives (in cm)
mean
sd
X1
5.84
0.83
Note that this is quite an overly complex way to return these summary statistics - using the tidyverse() way is much more intuitive and straightforward!
library(psych)library(kableExtra)# using the pre-loaded iris dataset# we want to get descriptive statistics of the iris dataset, specifically the sepal length column by Species# we want to return a matrix (hence mat = TRUE), then convert this to a dataframe# we specifically want to select the mean and standard deviation from the descriptive statistics available (try this without including this argument to see what values you all get out)# returning a table with a new column names of Group, Mean, SD; adding a caption; numbers are rounded to 2 dp# asking for a table that is not the full width of the window displaydescribeBy(Sepal.Length~Species, data =iris, mat =TRUE, digits =2)|>as.data.frame()|>rownames_to_column()|>select(group1, mean, sd)|>kable(col.names =c("Group", "Mean", "SD"), caption ="Sepal Length Descriptives (in cm)", digits =2)|>kable_styling(full_width =FALSE)
Sepal Length Descriptives (in cm)
Group
Mean
SD
setosa
5.01
0.35
versicolor
5.94
0.52
virginica
6.59
0.64
Correlation
Correlation Coefficient
The correlation coefficient - \(r_{(x,y)}=\frac{\mathrm{cov}(x,y)}{s_xs_y}\) - is a standardised number which quantifies the strength and direction of the linear association between two variables. In a population it is denoted by \(\rho\), and in a sample it is denoted by \(r\).
Values of \(r\) fall between \(-1\) and \(1\). How to interpret:
Size
More extreme values (i.e., the The closer \(r\) is to \(+/- 1\)) the stronger the linear association, and the closer to \(0\) a weak/no association. Commonly used cut-offs are:
Weak = \(.1 < |r| < .3\)
Moderate = \(.3 < |r| < .5\)
Strong = \(|r| > .5\)
Direction
The sign of \(r\) says nothing about the strength of the association, but its nature and direction:
Positive association means that values of one variable tend to be higher when values of the other variable are higher
Negative association means that values of one variable tend to be lower when values of the other variable are higher
Correlation Matrix
A correlation matrix is a table showing the correlation coefficients between variables. Each cell in the table shows the association between two variables. The diagonals show the correlation of a variable with itself (and are therefore always equal to 1).
In R
We can create a correlation matrix by giving the cor() function a dataframe. It is important to remember that all variables must be numeric. One way to check this is by using the str() argument.
Let’s check the structure of the iris dataset to ensure that all variables are numeric:
We can see that the variable Species in column 5 is a factor - this means that we cannot include this in our correlation matrix. Therefore, we need to subset, or, in other words, select specific columns. We can do this either giving the column numbers inside [], or using select(). In our case, we want the variables in columns 1 - 4, just not 5.
If you had NA values within your dataset, you could choose to remove these NAs using na.rm = TRUE inside the cor() function.
# select only the columns we want by variable name, and pass this to cor()iris|>select(Sepal.Length, Sepal.Width, Petal.Length, Petal.Width)|>cor()|>round(digits =2)
The hypotheses of the correlation test are, as always, statements about the population parameter (in this case the correlation between the two variables in the population - i.e., \(\rho\)).
If we are conducting a two tailed test, then…
\(H_0: \rho = 0\). There is no linear association between \(x\) and \(y\) in the population
\(H_1: \rho \neq 0\) There is a linear association between \(x\) and \(y\)
If we instead conduct a one-tailed test, then we are testing either…
\(H_0: \rho \leq 0\) There is a negative or no linear association between \(x\) and \(y\)
\(H_1: \rho > 0\) There is a positive linear association between \(x\) and \(y\)
OR
\(H_0: \rho \geq 0\) There is a positive or no linear association between \(x\) and \(y\)
\(H_1: \rho < 0\) There is a negative linear association between \(x\) and \(y\)
Test Statistic
The test statistic for this test is the \(t\) statistic, the formula for which depends on both the observed correlation (\(r\)) and the sample size (\(n\)):
\[t = r \sqrt{\frac{n-2}{1-r^2}}\]
p-value
We calculate the \(p\)-value for our \(t\)-statistic as the long-run probability of a \(t\)-statistic with \(n-2\) degrees of freedom being less than, greater than, or more extreme in either direction (depending on the direction of our alternative hypothesis) than our observed \(t\)-statistic.
Assumptions
For a test of Pearson’s correlation coefficient \(r\), we need to make sure a few conditions are met:
Both variables are quantitative (i.e., continuous)
Both variables are drawn from normally distributed populations
The association between the two variables is linear
No extreme outliers in dataset
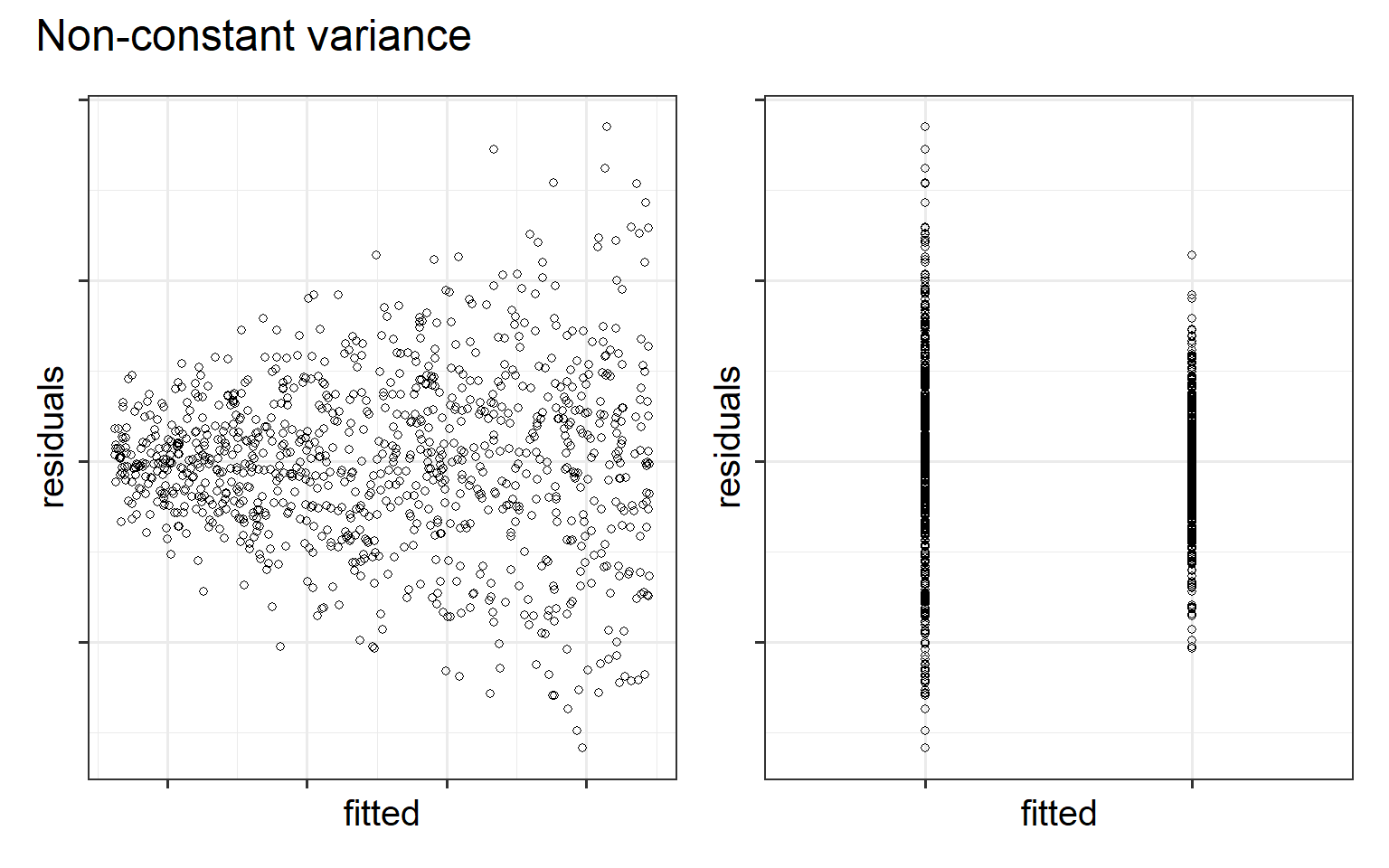
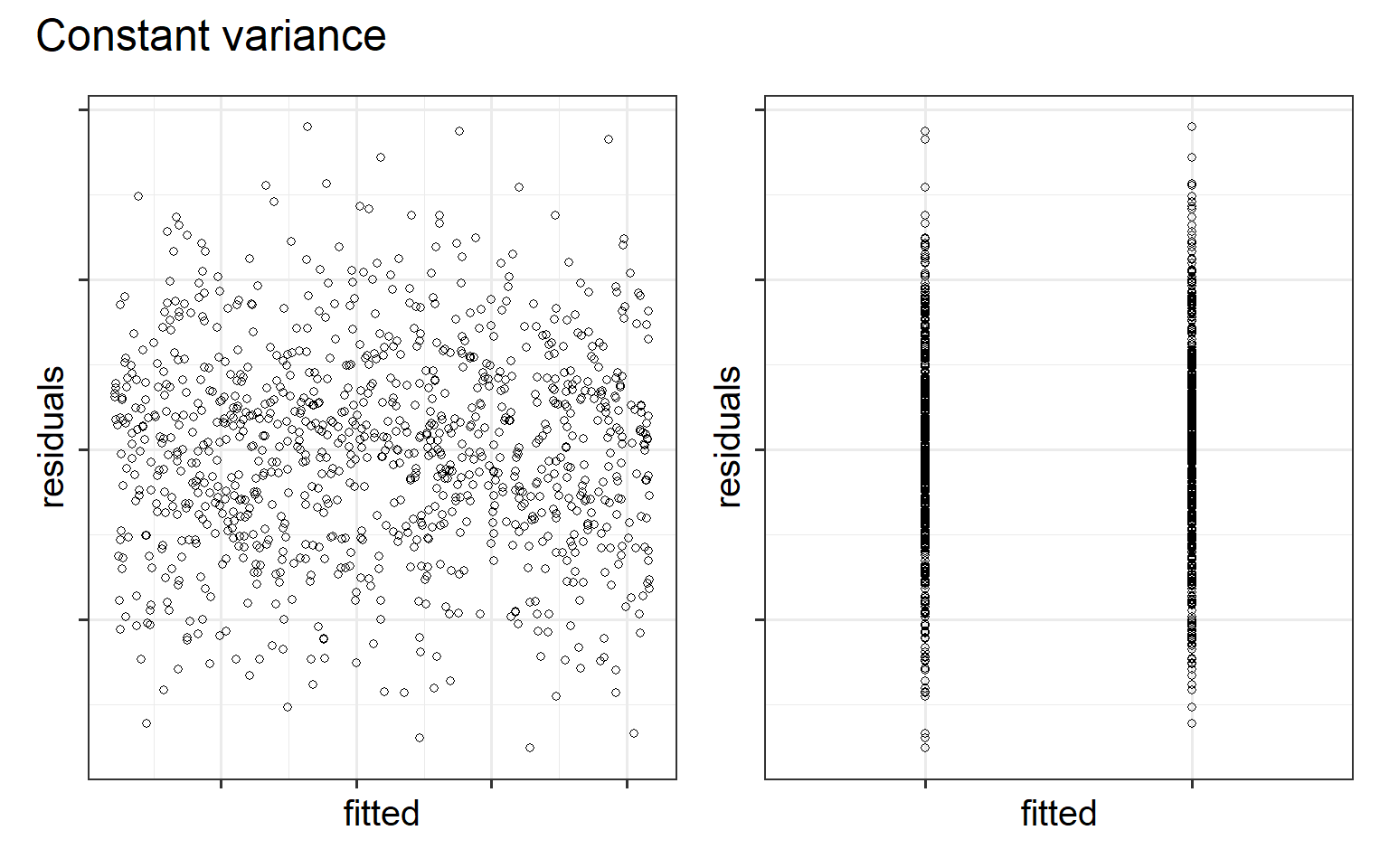
Homoscedasticity (homogeneity of variance)
Correlation - Hypothesis Testing in R
In R
We can test the significance of the correlation coefficient really easily with the function cor.test():
Pearson's product-moment correlation
data: iris$Sepal.Length and iris$Petal.Length
t = 21.646, df = 148, p-value < 2.2e-16
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.8270363 0.9055080
sample estimates:
cor
0.8717538
Note, by default, cor.test() will include only observations that have no missing data on either variable.
We can specify whether we want to conduct a one- or two-tailed test by adding the argument alternative = and specifying alternative = "less", alternative = "greater", or alternative = "two.sided" (the latter being the default).
Example Interpretation
There was a strong positive association between sepal length and petal length \((r = .87, t(148) = 21.65, p < .001)\). These results suggested that a greater sepal length was positively associated with a greater petal length.
Note
For a detailed recap of all things correlation (including further details and examples), revisit the Correlation lecture from DAPR1.
Visual Exploration
Visual exploration of our data allows us to visualize the distributions of our data, and to identify potential associations among variables.
How to Visualise Data
To visualise (i.e., plot) our data, we can use ggplot() from the tidyverse package. Note the key components of the ggplot() code:
data = where we provide the name of the dataframe
aes = where we provide the aesthetics. These are things which we map from the data to the graph. For instance, the \(x\)-axis, or if we wanted to colour the columns/bars according to some aspect of the data
+ geom_... = where we add (using +) some geometry. These are the shapes (e.g., bars, points, etc.), which will be put in the correct place according to what we specified in aes()
labs() = where we provide labels for our plot (e.g., the \(x\)- and \(y\)-axis)
Note
There are lots of arguments that you can further customise, some of which are specified in the examples below e.g., bins =, alpha =, fill =, linewidth =. linetype =, size = etc. For these, you can look up the helper function to see the range of arguments they can take using ? - e.g., ?fill.
One other thing to consider when visualising your data is how you are going to arrange your plots. Some handy tips on this:
Use to wrap text in your titles and or axis labels
The patchwork package allows us to arrange multiple plots in two ways - | arranges the plots adjacent to one another, and / arranges the plots on top of one another
“Density” is a bit similar to the notion of “relative frequency” (or “proportion”), in that for a density curve, the values on the y-axis are scaled so that the total area under the curve is equal to 1. In creating a curve for which the total area underneath is equal to one, we can use the area under the curve in a range of values to indicate the proportion of values in that range.
Unlike in our marginal plots where we specified our x-axis variable within aes(), to visualise bivariate associations, we need to specify what variables we want on both our x- and y-axis.
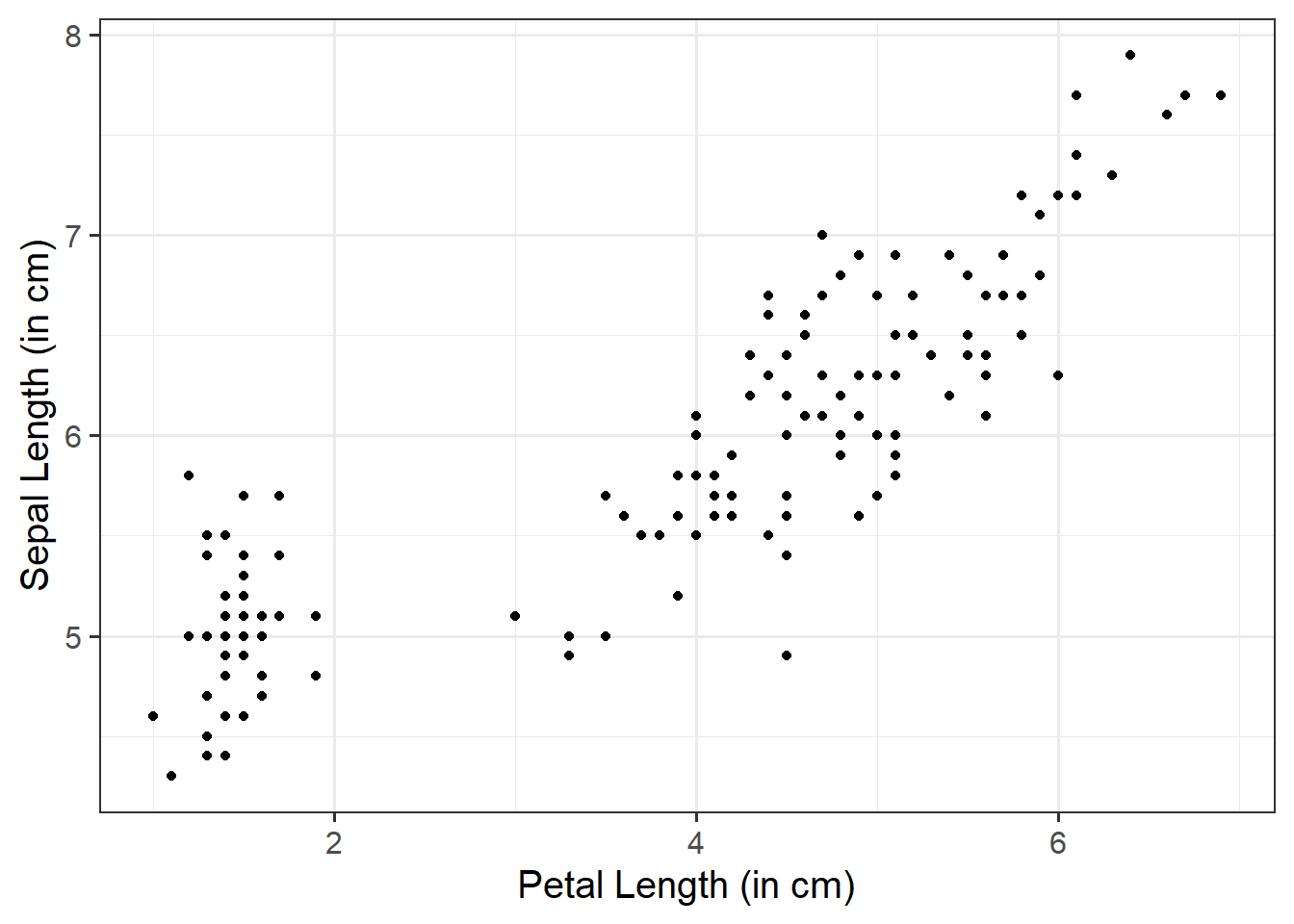
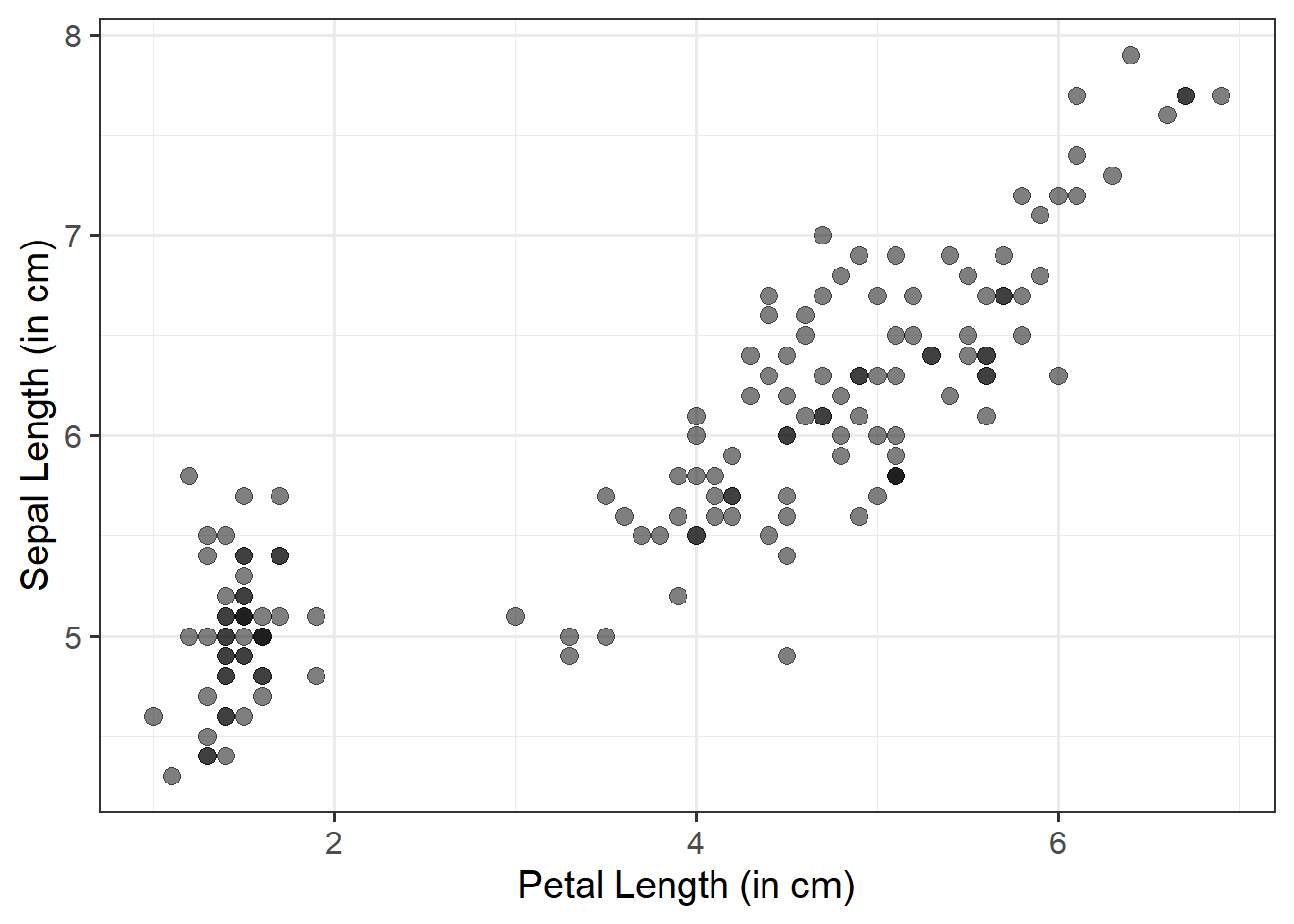
We can use a scatterplot (since the variables are numeric and continuous) to visualise the association between the two numeric variables - these will be our x- and y-axis values.
We plot these values for each row of our dataset, and we should end up with a cloud of scattered points.
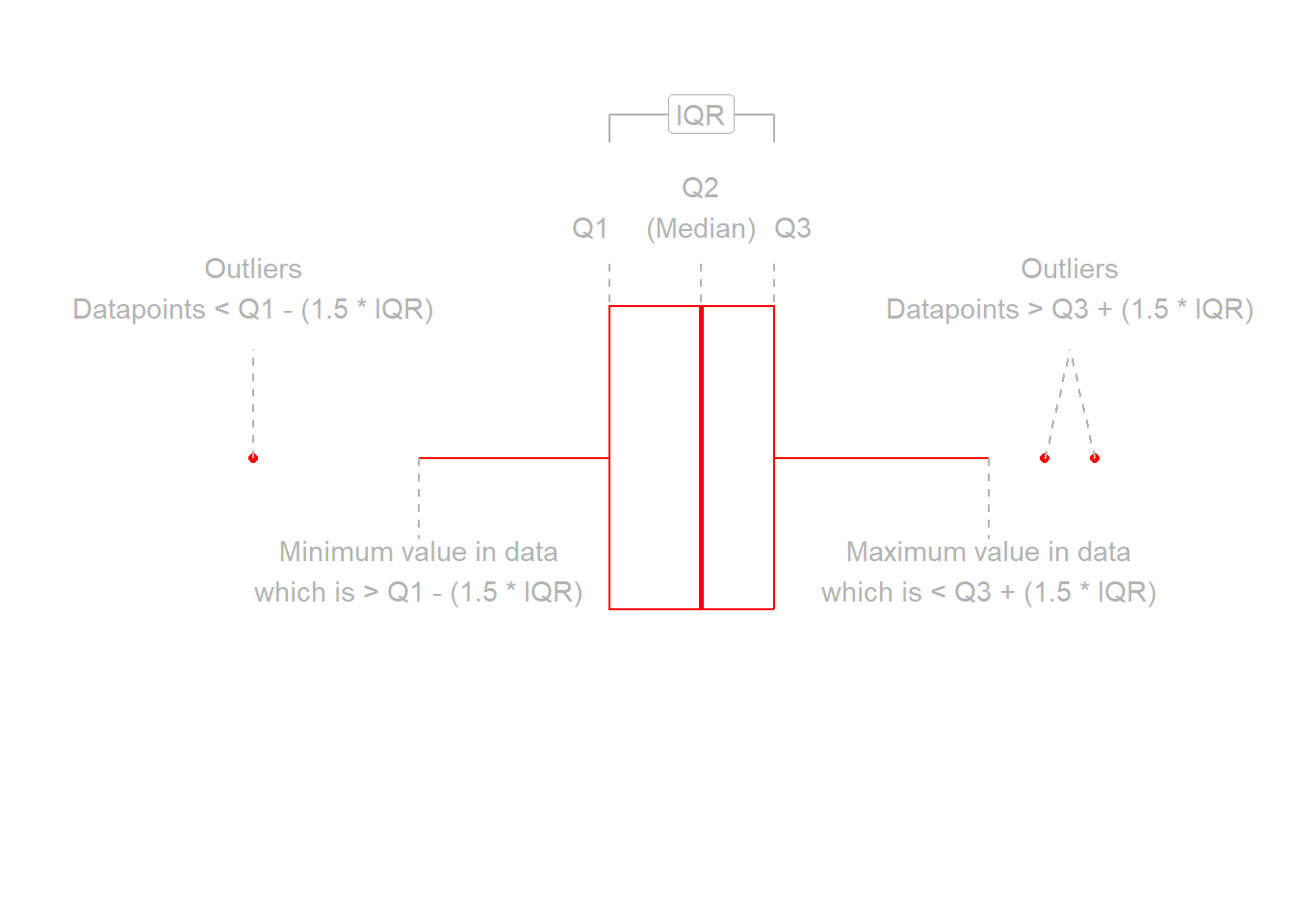
Here we will want to comment on any key observations that we notice, including if we detect outliers or points that do not fit with the pattern in the rest of the data. Outliers are extreme observations that are not possible values of a variable or that do not seem to fit with the rest of the data. This could either be:
marginally along one axis: points that have an unusual (too high or too low) x-coordinate or y-coordinate
jointly: observations that do not fit with the rest of the point cloud
Basic:
We need to specify + geom_point() to get a scatterplot:
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point()+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
Fill points with color:
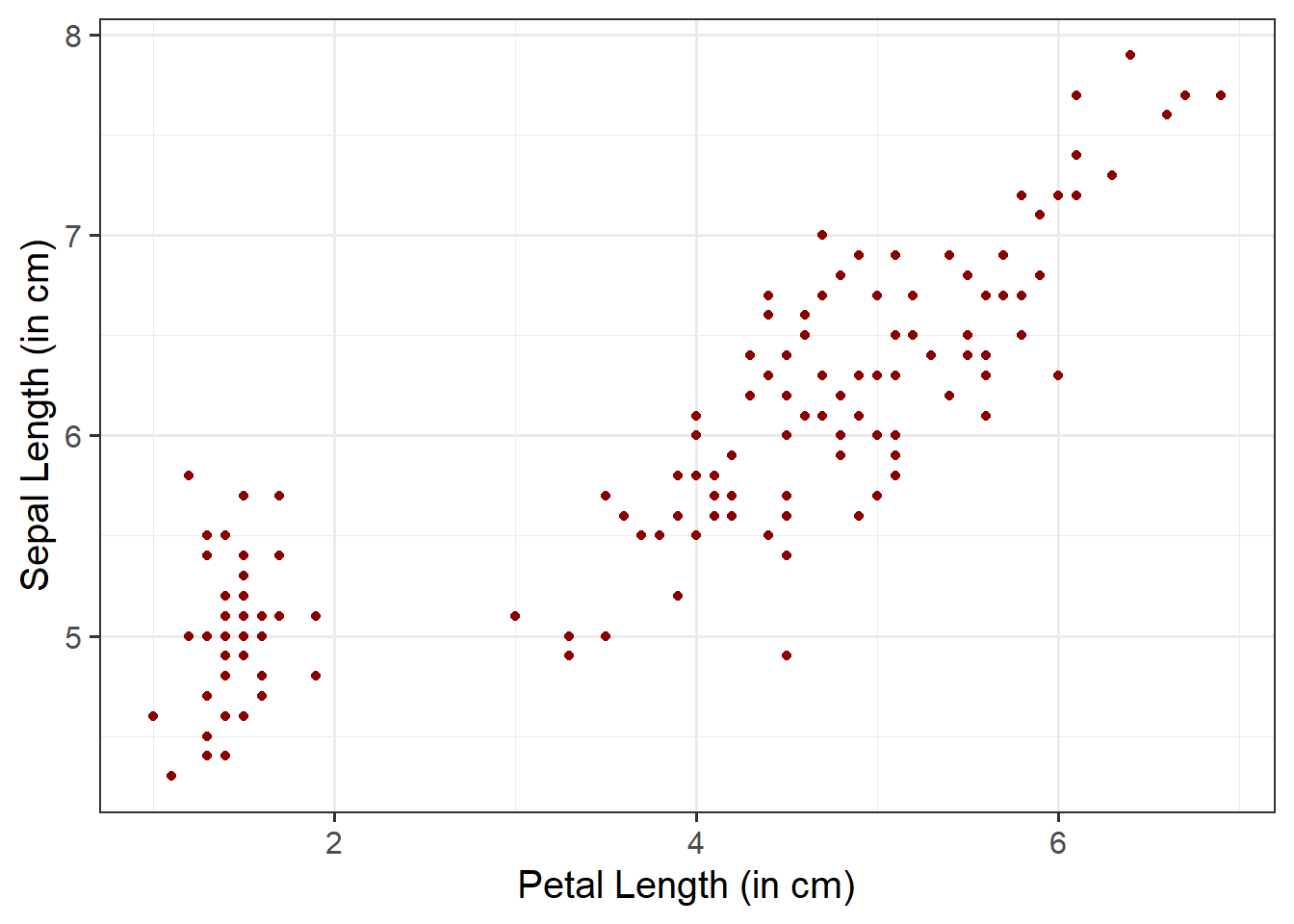
Within geom_point(), we can specify color = to fill the points with a color:
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point(color ="darkred")+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
Change size and opacity:
We can change the size (using size =) and the opacity (using alpha =) of our geom elements on the plot. Let’s include this below:
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point(size =3, alpha =0.5)+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
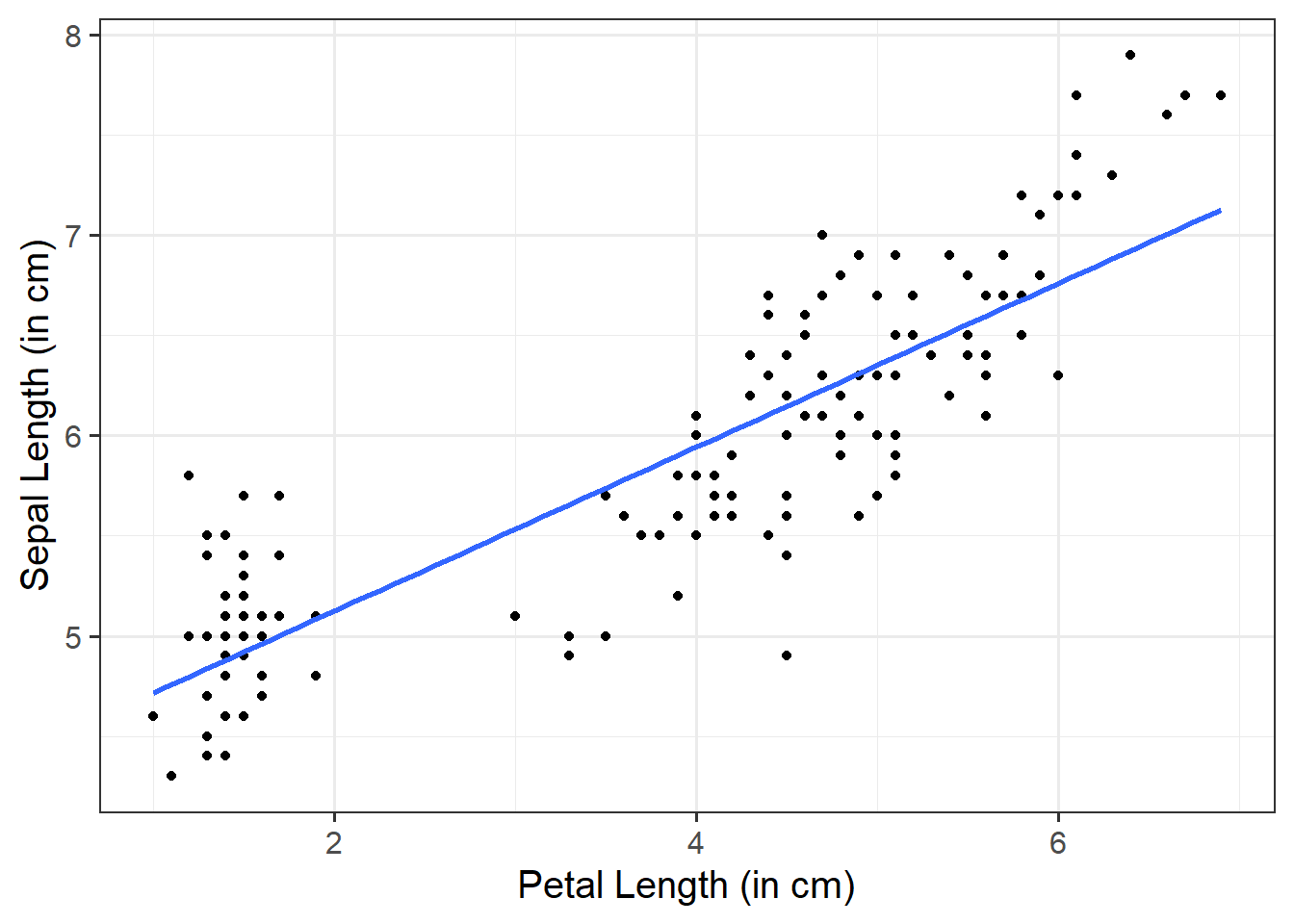
Add a line of best fit:
We can superimpose (i.e., add) a line of best fit by including the argument + geom_smooth(). Since we want to fit a straight line, we want to use method = "lm". We can also specify whether we want to display confidence intervals around our line by specifying se = TRUE / FALSE.
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point()+geom_smooth(method ="lm", se =FALSE)+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
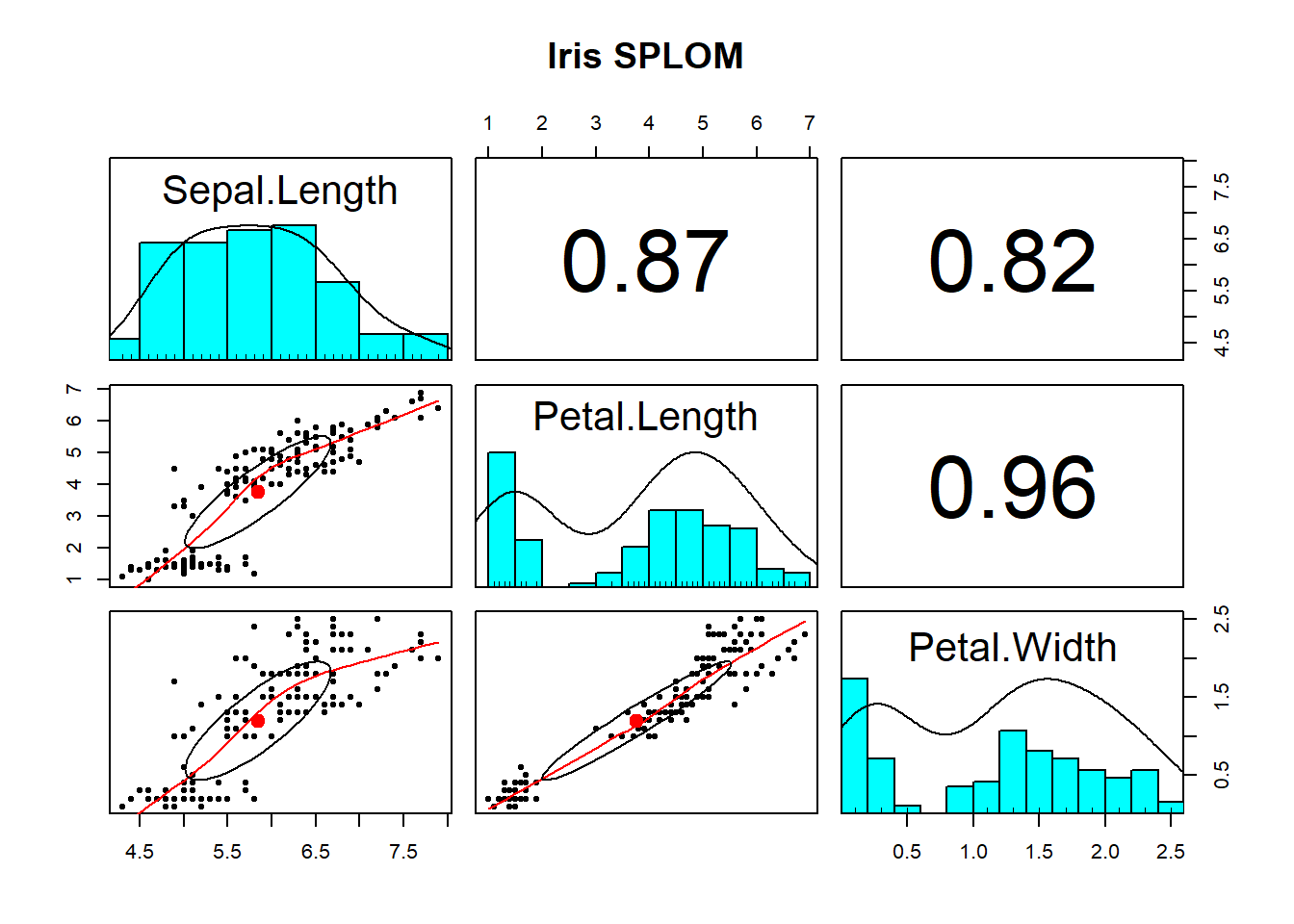
Using pairs.panels() is likely the most useful way to visualise the associations among numeric variables. It returns a scatterplot of matrices (SPLOM) returning you (1) the marginal distribution of each variable via a histogram, (2) the correlation between variables, and (3) bivariate scatterplots.
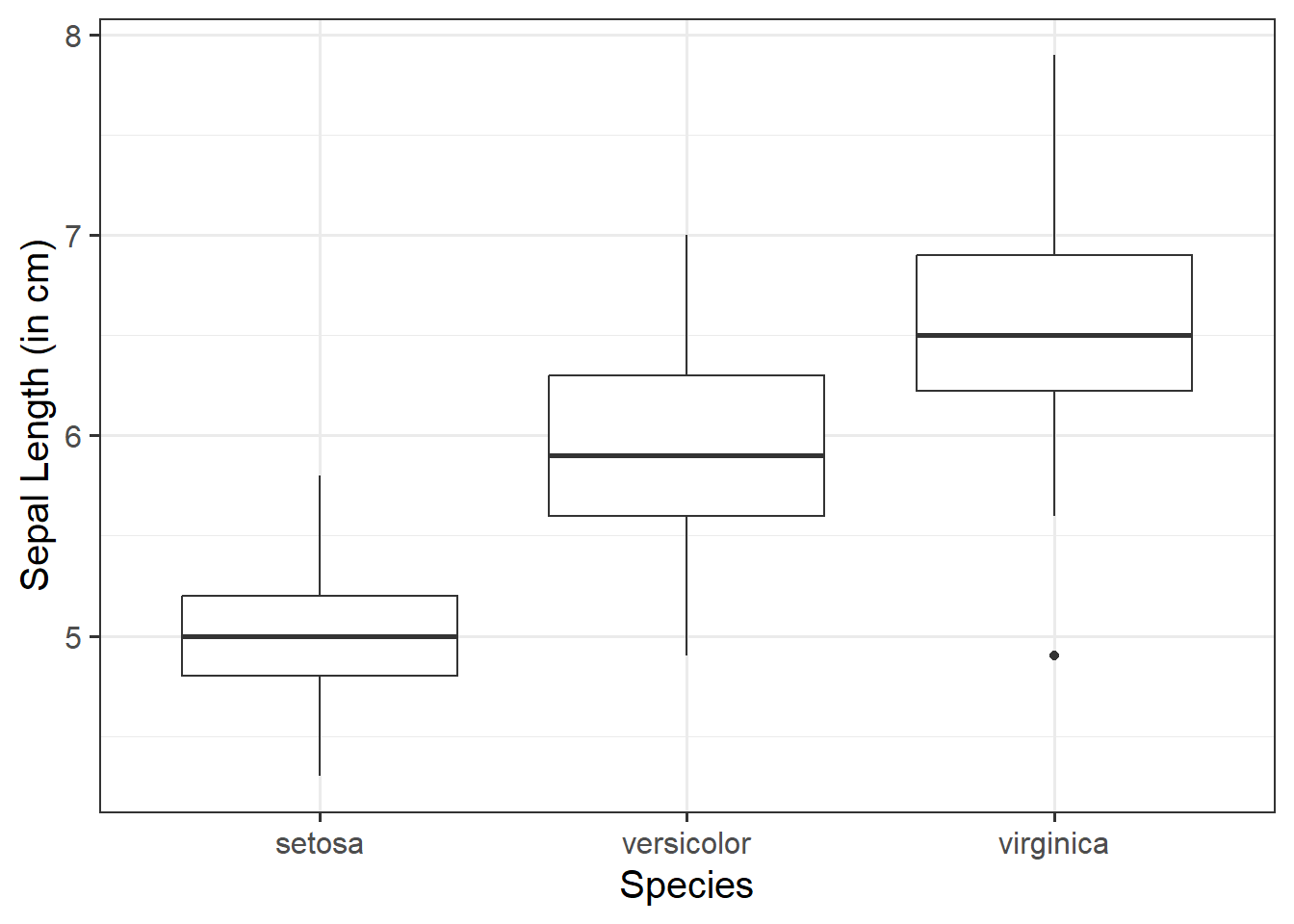
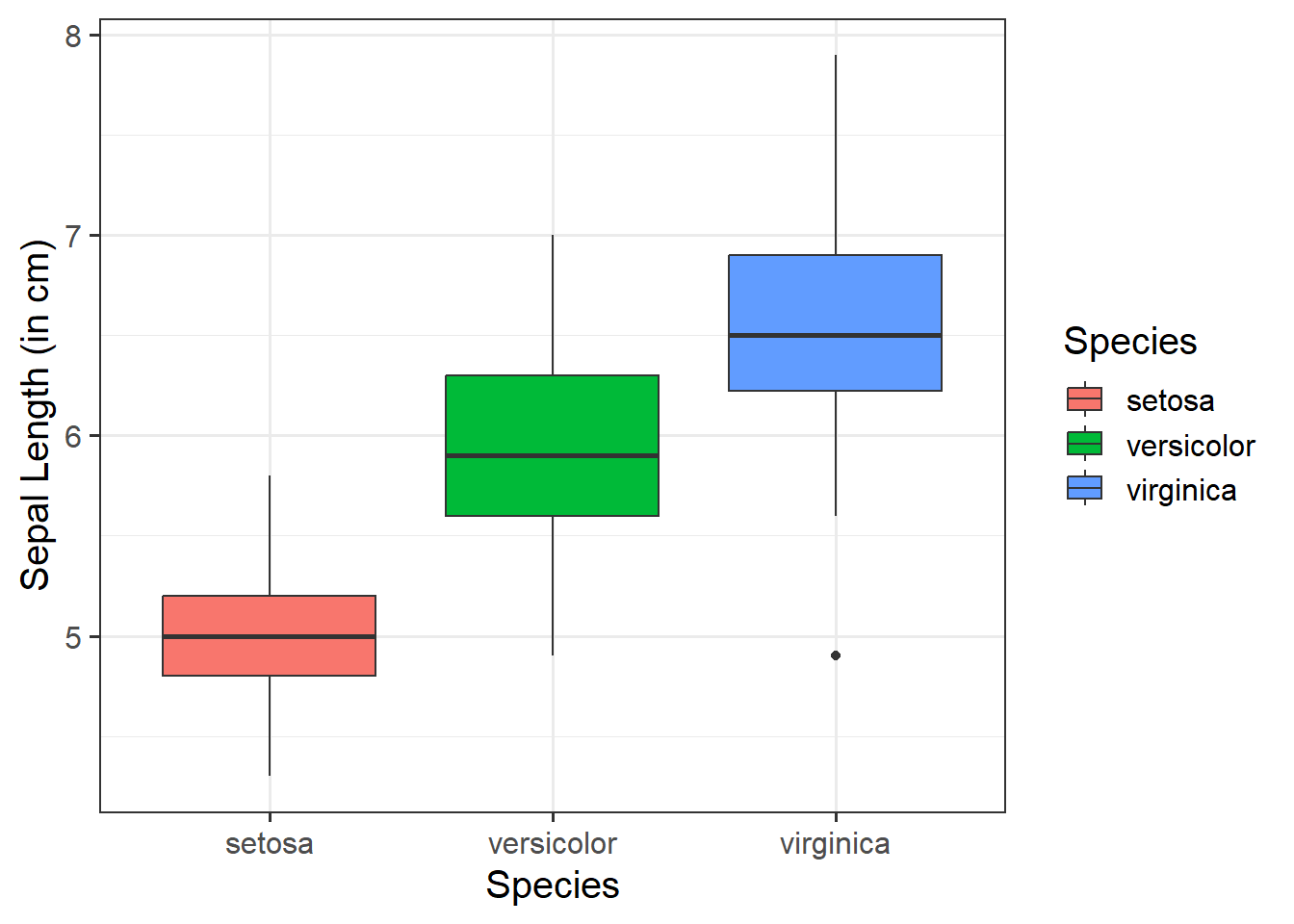
We can use a boxplot to visualise the association between one numeric variable and one categorical variable - these will be our y- and x-axis values respectively. This can be helpful to visually compare the distribution of multiple groups.
Basic:
We need to specify + geom_boxplot() to get a boxplot:
ggplot(data =iris, aes(x =Species, y =Sepal.Length))+geom_boxplot()+labs(x ="Species", y ="Sepal Length (in cm)")
Change boxplot fill colours by group:
Within aes(), we can specify fill = to fill the boxes with a color:
ggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_boxplot()+labs(x ="Species", y ="Sepal Length (in cm)")
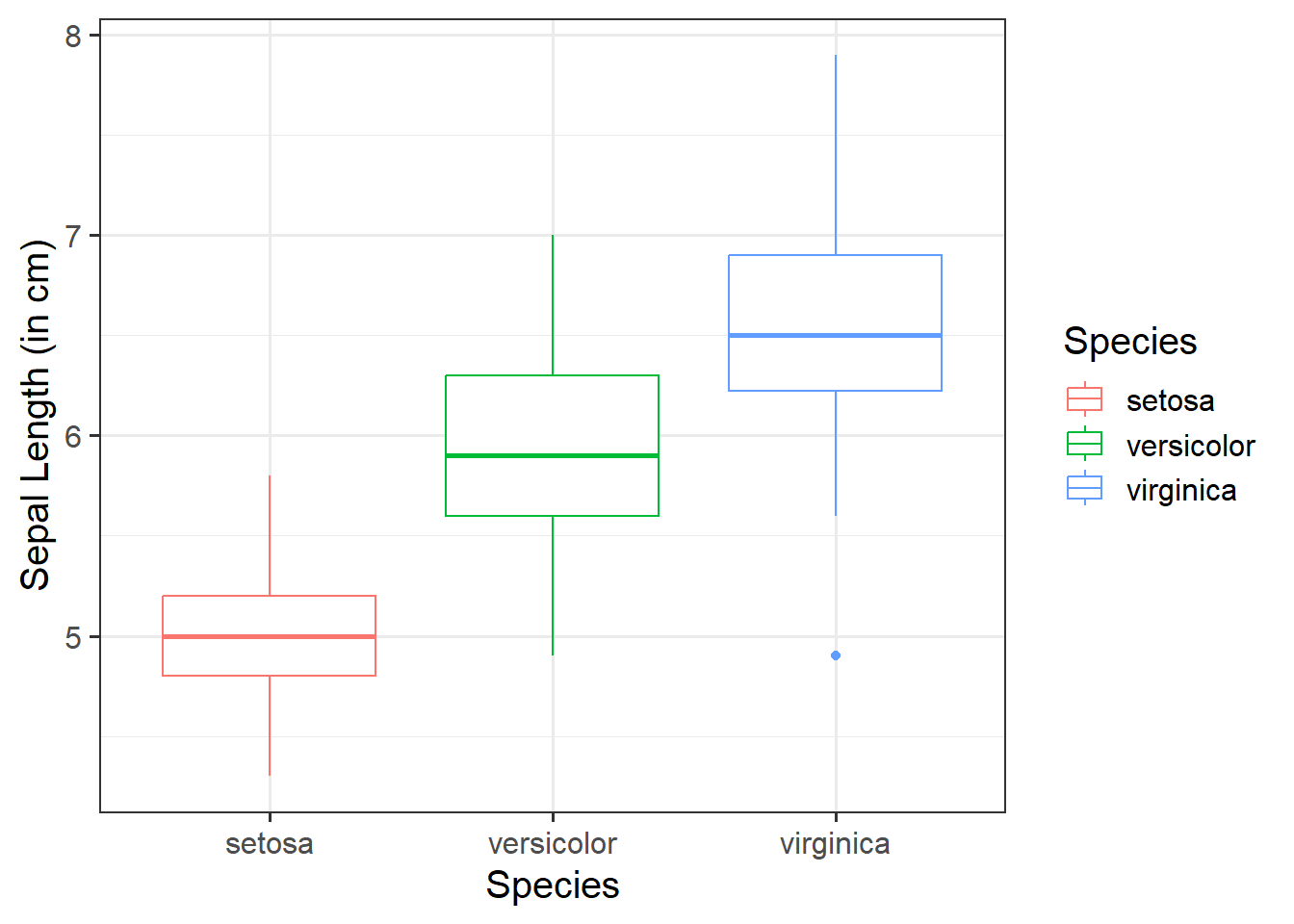
Change boxplot line colours by group:
Within aes(), we can specify color = to colour the lines with a color:
ggplot(data =iris, aes(x =Species, y =Sepal.Length, color =Species))+geom_boxplot()+labs(x ="Species", y ="Sepal Length (in cm)")
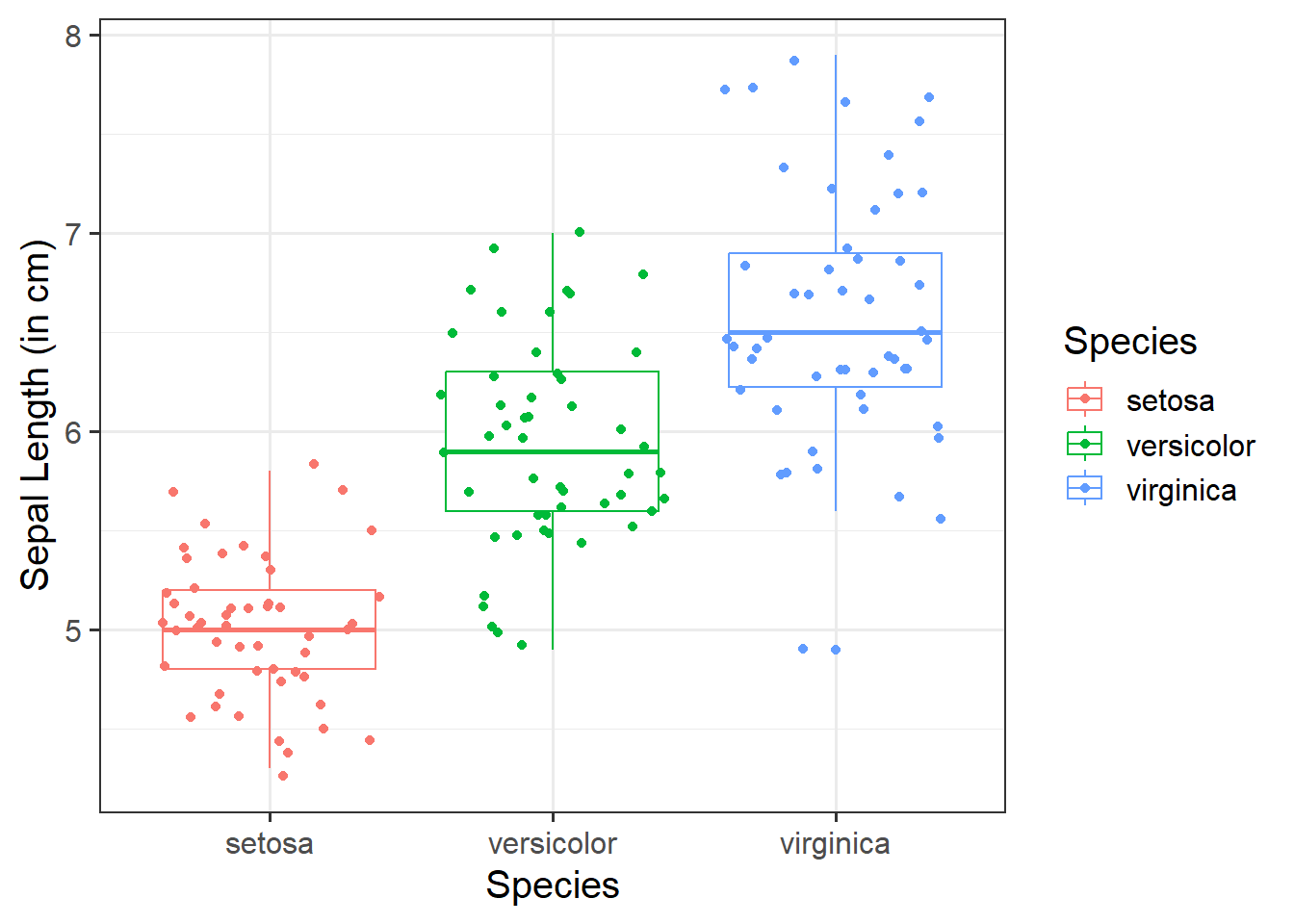
Adding jitter:
We can add jittered points to a boxplot to better see the underlying distribution of the data (by adding a little random variation to each data point) via geom_jitter():
We can add the argument + theme(legend.position = ) to move (or even remove) the legend by specifying, for example, "right", "left", "top", "bottom", or "none" to remove.
# legend at bottom of plotggplot(data =iris, aes(x =Species, y =Sepal.Length, color =Species))+geom_boxplot()+labs(x ="Species", y ="Sepal Length (in cm)")+theme(legend.position ="bottom")
Like a boxplot, we can use a violin plot to visualise the association between one numeric variable and one categorical variable - these will be our y- and x-axis values respectively. It combines a summary of the data’s range and a kernel density estimation, providing a detailed view of the distribution.
Basic:
We need to specify + geom_violin() to get a violin plot:
ggplot(data =iris, aes(x =Species, y =Sepal.Length))+geom_violin()+labs(x ="Species", y ="Sepal Length (in cm)")
Change violin colours by group:
Within aes(), we can specify fill = to fill the violins with a color:
ggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_violin()+labs(x ="Species", y ="Sepal Length (in cm)")
Change Size and opacity:
We can change the size (using size =) and the opacity (using alpha =) of our geom elements on the plot. Let’s include this below:
ggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_violin(size =1, alpha =0.5)+labs(x ="Species", y ="Sepal Length (in cm)")
Adding jitter:
We can add jittered points to a violin plot to better see the underlying distribution of the data (by adding a little random variation to each data point) via geom_jitter():
ggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_violin()+geom_jitter(alpha =0.5)+labs(x ="Species", y ="Sepal Length (in cm)")
Change legend position:
We can add the argument + theme(legend.position = ) to move (or even remove) the legend by specifying, for example, "right", "left", "top", "bottom", or "none" to remove.
# no legendggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_violin()+geom_jitter(alpha =0.5)+theme(legend.position ='none')+labs(x ="Species", y ="Sepal Length (in cm)")
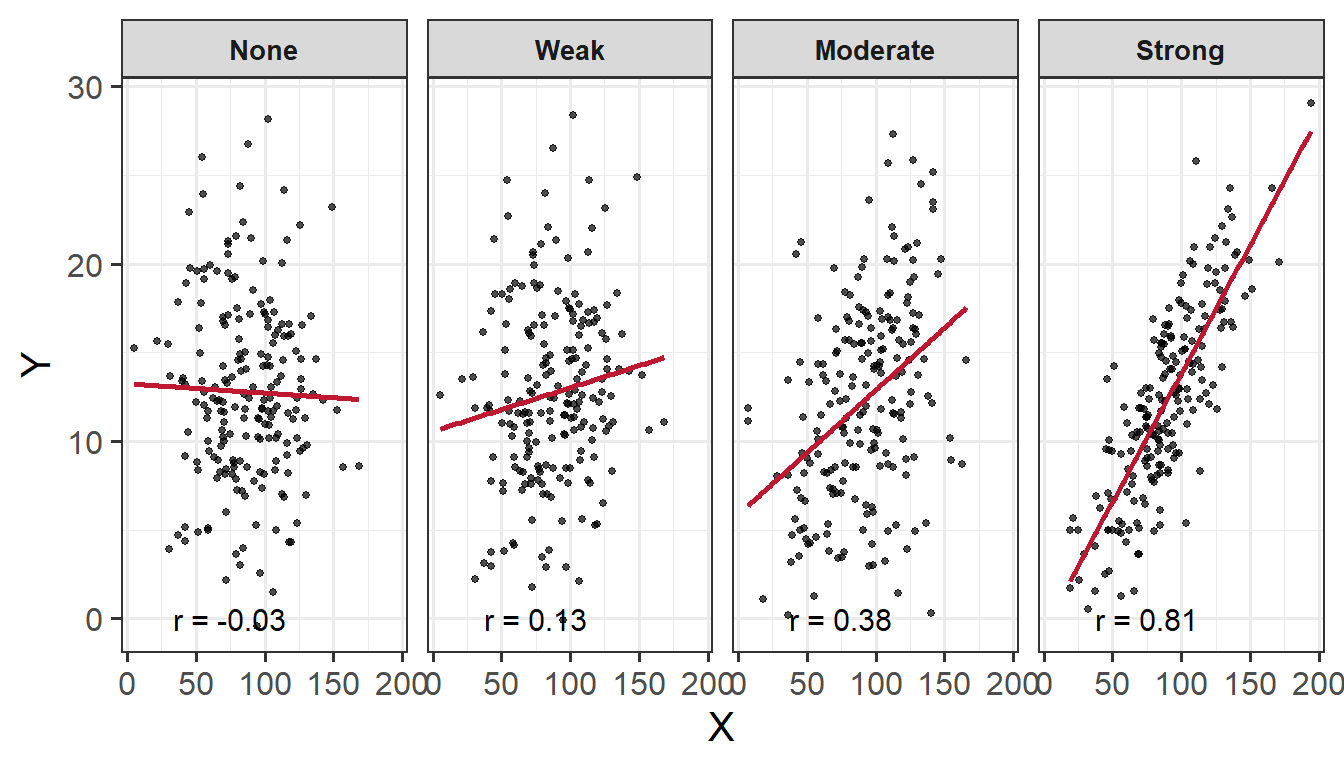
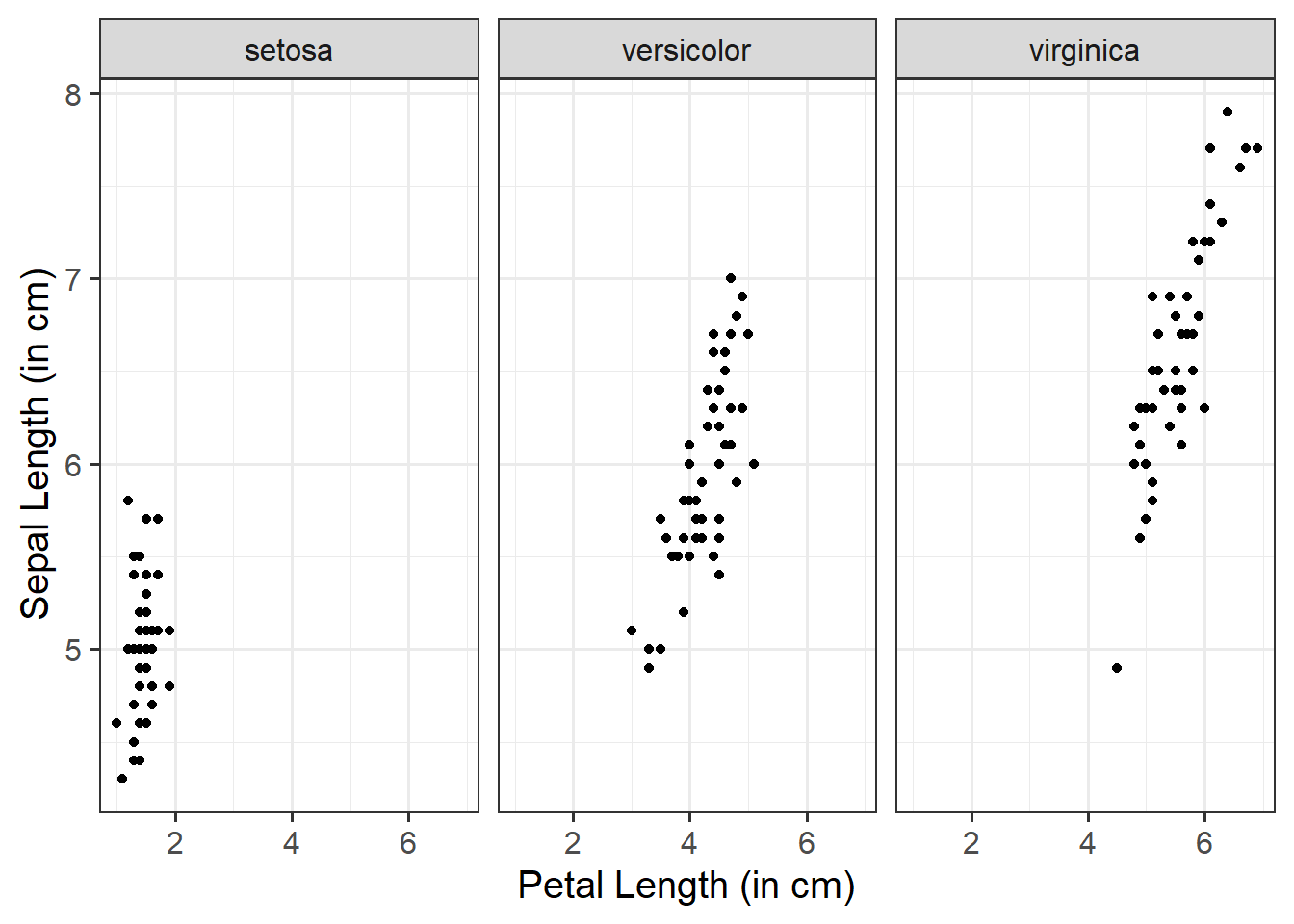
When we have two numeric variables, as well as categorical variables, we can use facet_wrap() / facet_grid() to help divide/arrange our plots. If we had two categorical variables, by simply stringing them together to further group our plots by specifying facet_wrap( ~ cat_variable1 + cat_variable2)
Basic:
We need to specify + geom_point() to get a scatterplot, and either+ facet_wrap() or + facet_grid() to separate by your categorical variable:
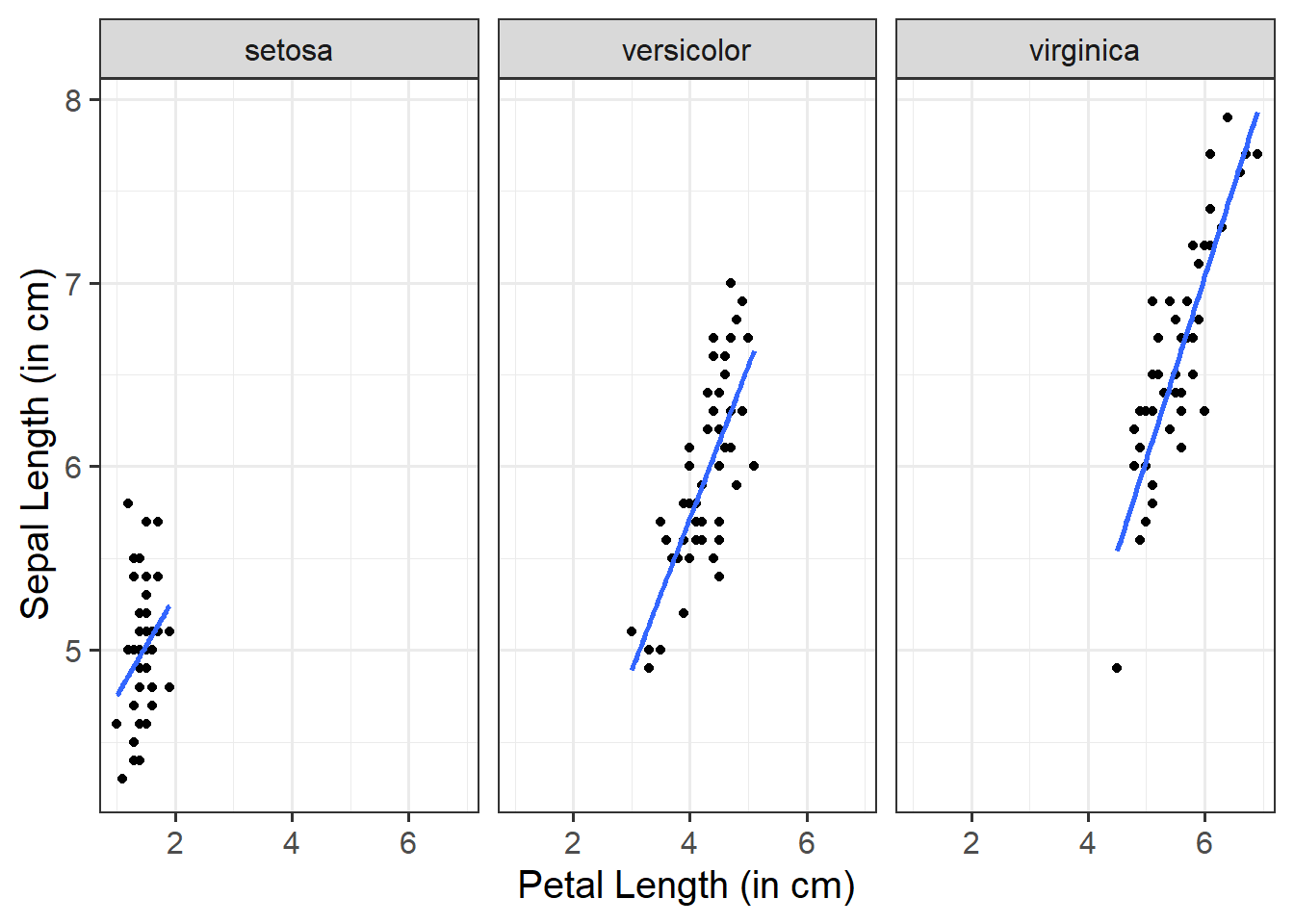
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point()+facet_wrap(~Species)+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
Add a line of best fit:
We can superimpose (i.e., add) a line of best fit by including the argument + geom_smooth(). Since we want to fit a straight line, we want to use method = "lm". We can also specify whether we want to display confidence intervals around our line by specifying se = TRUE / FALSE. Note that a line is fitted for every level of your categorical variable:
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point()+geom_smooth(method ="lm", se =FALSE)+facet_wrap(~Species)+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
Subplot layout:
You can change the overall layout of the subplots by specifying dir = within the facet_wrap() argument, where “h” will return a horizontal layout (this is the default) and “v” for vertical.
You can also change the layout of the subplot labels by specifying strip.position = within the facet_wrap() argument, where labels can be arranged to display at the “top” (this is the default), “bottom”, “left” or “right”.
ggplot(data =iris, aes(x =Petal.Length, y =Sepal.Length))+geom_point()+facet_wrap(~Species, dir ="v", strip.position ="right")+labs(x ="Petal Length (in cm)", y ="Sepal Length (in cm)")
Multivariate Associations - Examples
To visualise multivariate associations, just like we do for bivariate associations, we need to specify what variables we want on both our x- and y-axis. We also need to take an extra step by specifying a third variable - z - that acts as a differentiating factor across our data. This ‘z’ can be mapped to an aesthetic attribute such as color, shape, or size, allowing us to explore more dynamic patterns and ssociations in our data.
If you really wanted to, you could create a plot showing the associations among three variables at once. These are likely more useful when you have an interaction model. However, we wouldn’t really recommend doing this - they can be very difficult to interpret correctly, and given their interactive nature, definitely NOT something that you’d want to include in a stats report. But, for demonstration purposes only, we could create one using the plotly package.
3D Scatterplot
library(plotly)plot_ly(data =iris, x =~Petal.Length, y =~Sepal.Length, z =~Petal.Width, type ='scatter3d', mode ='markers+lines', scene =list( xaxis =list(title ="Petal Length"), yaxis =list(title ="Sepal Length"), zaxis =list(title ="Petal Width")))
Heatmap of Correlations
plot_ly(z =~cor(iris[, c(1, 3:4)]), type ="heatmap")
Functions and Mathematical Models
Basic functions and mathematical models are foundational tools used to describe and predict associations between variables.
Identification & Specification
Consider the function \(y = 2 + 5 \ x\). From this, we can do the following:
Identify the Dependent Variable (DV)
Identify the Independent Variable (IV)
Describe in words what the function does, and compute the output for the following input:
\[
x = \begin{bmatrix}
2 \\
6
\end{bmatrix}
\]
The function says that the \(y\) value is obtained as a transformation of the \(x\) value.
The dependent variable is \(y\)
The independent variable is \(x\)
The \(y\) value is calculated as two plus five times \(x\)
Example (1): If \(x\) equals 2, the corresponding value of \(y\) will be \(2 + (5 \cdot 2) = 12\). Example (2): If \(x\) equals 6, the corresponding value of \(y\) will be \(2 + (5 \cdot 6) = 32\).
We come across functions a lot in daily life, and probably don’t think much about it. In a slightly more mathematical setting, we can write down in words and in symbols the function describing the association between the side of a square and its perimeter (e.g., to capture how the perimeter varies as a function of its side). In this case, the perimeter is the dependent variable, and the side is the independent variable.
This is what we would refer to as a deterministic model, as it is a model of an exact relationship - there can be no deviation.
The perimeter of a square is four times the length of its side.
The relationship between side and perimeter of squares is given by:
\[
\text{Perimeter} = 4 \cdot \text{Side}
\]
If you denote \(y\) as the dependent variable Perimeter, and \(x\) as the independent variable Side we can rewrite as:
\[
y = 4 \cdot x
\]
Visualisation
Let’s create a dataset called squares, containing the perimeter of four squares having sides of length \(0, 2, 5, 9\) metres, and then plot the squares data as points on a scatterplot.
First, let’s make our squares data. Here we will use two important functions - tibble() and c(). The tibble() function allows us to construct a data frame. To store a sequence of numbers into R, we can combine the values using c(). A sequence of elements all of the same type is called a vector.
#create data frame named squaressquares<-tibble( side =c(0, 2, 5, 9), perimeter =4*side)#check that our values are contained within squaressquares
Now we know how ggplot() works, we can start to build our plot. First we specify our data (we want to use the squares data frame), and then our aesthetics. Since the perimeter varies as a function of side, we want side on the \(x\)-axis, and perimeter on the \(y\)-axis. We want to create a scatterplot, so we need to specify our geom_... argument as geom_point(). Lastly, we will provide clearer axis labels, and include the units of measurement.
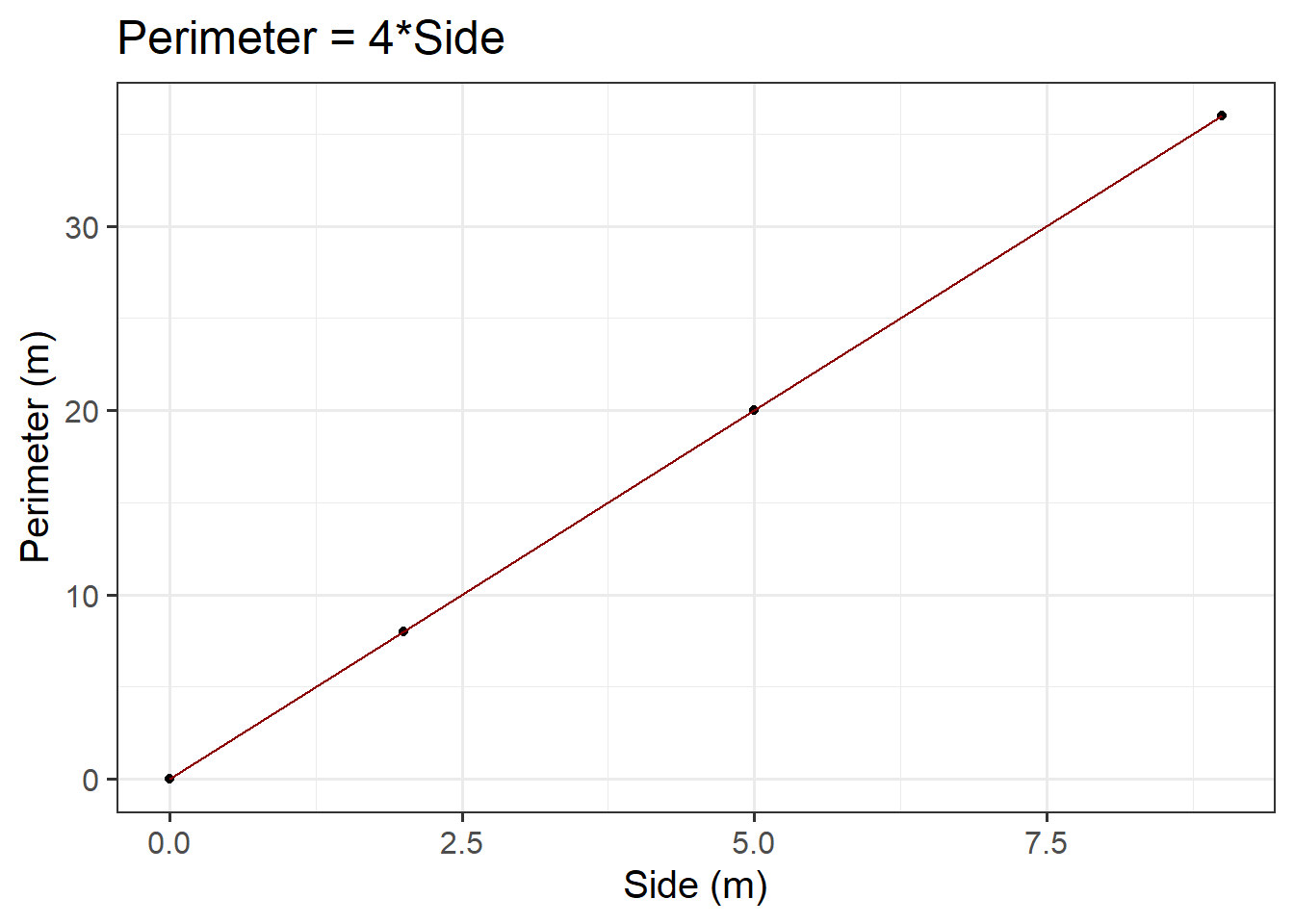
ggplot(data =squares, aes(x =side, y =perimeter))+geom_point()+labs(x ='Side (m)', y ='Perimeter (m)', title ='Perimeter = 4*Side')
Figure 3: Perimeter = 4*Side
We could also visualise the functional relationship by connecting the individual points with a line. To do so, we need to add a new argument - geom_line(). If you would like to change the colour of the line from the default, you can specify geom_line(colour = "insert colour name").
ggplot(data =squares, aes(x =side, y =perimeter))+geom_point()+geom_line(colour ="darkred")+labs(x ='Side (m)', y ='Perimeter (m)', title ='Perimeter = 4*Side')
Figure 4: Perimeter = 4*Side
Predicted Values
Sometimes we can directly read a predicted value from the graph of the functional relationship.
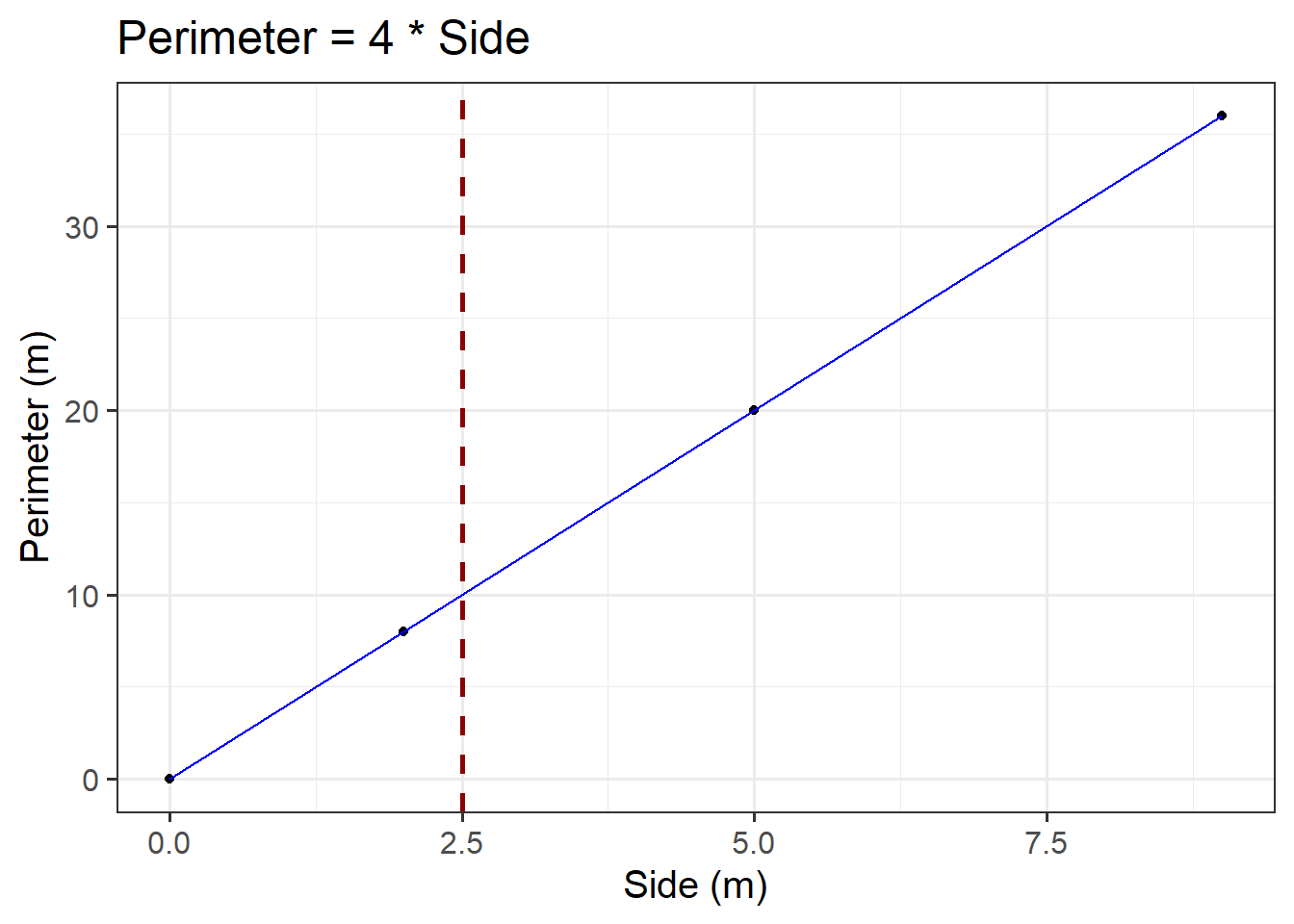
Consider the plot created above. For example, first we need to check where \(x\) = 2.5. Then, we draw a vertical dashed line until it meets the blue line. The \(y\) value corresponding to \(x\) = 2.5 can be read off the \(y\)-axis. In our case, we would say a side of 2.5m corresponds to a perimeter of 10m.
ggplot(data =squares, aes(x =side, y =perimeter))+geom_point()+geom_line(colour ="blue")+geom_vline(xintercept =2.5, colour ="darkred", lty ="dashed", lwd =1)+labs(x ='Side (m)', y ='Perimeter (m)', title ='Perimeter = 4 * Side')
Figure 5: Perimeter = 4*Side
However, in this case it is not that easy to read it from Figure 5 (especially without the superimposed dashed red line)… This leads us to the algebraic approach:
We can substitute the \(x\) value in the formula and calculate the corresponding \(y\) value where we would conclude that the predicted perimeter of squared paintings having a 2.5m side is 10m:
\[
y = 4 \cdot x \\
\]
\[
y = 4 \cdot 2.5 \\
\]
\[
y = 10 \\
\]
Statistical Models
Statistical models are used to understand the associations among variables.
Specifying Hypotheses
We need to specify our hypotheses when testing a model as this not only defines what we are testing, but also sets the direction for statistical inference. By specifying a null hypothesis (typically stating no effect or no association) and an alternative hypothesis (indicating the presence of an association), we create a structured approach for determining the statistical significance of model parameters. Without specifying hypotheses, the interpretation of results would lack focus, making it difficult to assess the validity and relevance of the model’s findings.
In regression analysis, hypothesis testing for beta coefficients is used to assess whether (each) predictor variable significantly contributes to the model.
The way we specify hypotheses is similar across simple and multiple regression models.
For each regression coefficient \(\beta_j\) (for predictor \(X_j\)):
Null hypothesis (\(H_0\)) = \(\beta_j = 0\): The predictor variable (\(X_j\)) is not associated with the DV
Alternative hypothesis (\(H_1\)) = \(\beta_j \neq 0\): The predictor variable (\(X_j\)) is associated with the DV
Based on the \(p\)-value or comparison of the \(t\)-statistic with the critical value, you can conclude whether the predictor variable is significant or not (see the simple & multiple regression Models - extracting information > model coefficients flashcard below):
Reject \(H_0\) if \(|t_j|\) > critical value or \(p\)-value \(< \alpha\)
Fail to reject \(H_0\) if \(|t_j|\)\(\leq\) critical value or \(p\)-value \(\geq \alpha\)
Numeric Outcomes & Numeric Predictors
Simple Linear Regression Models
Description & Model Specification
The association between two variables (e.g., recall accuracy and age) will show deviations from the ‘average pattern’. Hence, we need to create a model that allows for deviations from the linear relationship - we need a statistical model.
A statistical model includes both a deterministic function and a random error term. We typically refer to the outcome (‘dependent’) variable with the letter \(y\) and to our predictor (‘explanatory’/‘independent’) variables with the letter \(x\). A simple (i.e., one x variable only) linear regression model thus takes the following form (where the terms \(\beta_0\) and \(\beta_1\) are numbers specifying where the line going through the data meets the y-axis (i.e., the intercept - where \(x\) = 0; \(\beta_0\)) and its slope (direction and gradient of line; \(\beta_1\)):
\(N(0, \sigma) \text{ independently}\) means ‘normal distribution with a mean of 0 and a variance of \(\sigma\)’
Together, we can say that the errors around the line have a mean of zero and constant spread as x varies
In R
There are basically two pieces of information that we need to pass to the lm() function:
The formula: The regression formula should be specified in the form y ~ x where \(y\) is the dependent variable (DV) and \(x\) the independent variable (IV).
The data: Specify which dataframe contains the variables specified in the formula.
In R, the syntax of the lm() function can be specified as follows (where DV = dependent variable, IV = independent variable, and data_name = the name of your dataset):
When we specify the linear model in R, we include after the tilde sign (\(\sim\)), the variables that appear to the right of the \(\hat \beta\)s. The intercept, or \(\beta_0\), is a constant. That is, we could write it as multiplied by 1.
Including the 1 explicitly is not necessary because it is included by default (you can check this by comparing the outputs of A & B above with and without the 1 included - the estimates are the same!). After a while, you will find you just want to drop the 1 when calling lm() because you know that it’s going to be there, but in these early weeks we tried to keep it explicit to make it clear that you want the intercept to be estimated.
Example
Research Question
Is there an association between recall accuracy and age?
Overview
Imagine that you were tasked to investigate whether there was an association between recall accuracy and age. You have been provided with data from twenty participants who studied passages of text (c500 words long), and were tested a week later. The testing phase presented participants with 100 statements about the text. They had to answer whether each statement was true or false, as well as rate their confidence in each answer (on a sliding scale from 0 to 100). The dataset contains, for each participant, the percentage of items correctly answered, their age (in years), and their average confidence rating.
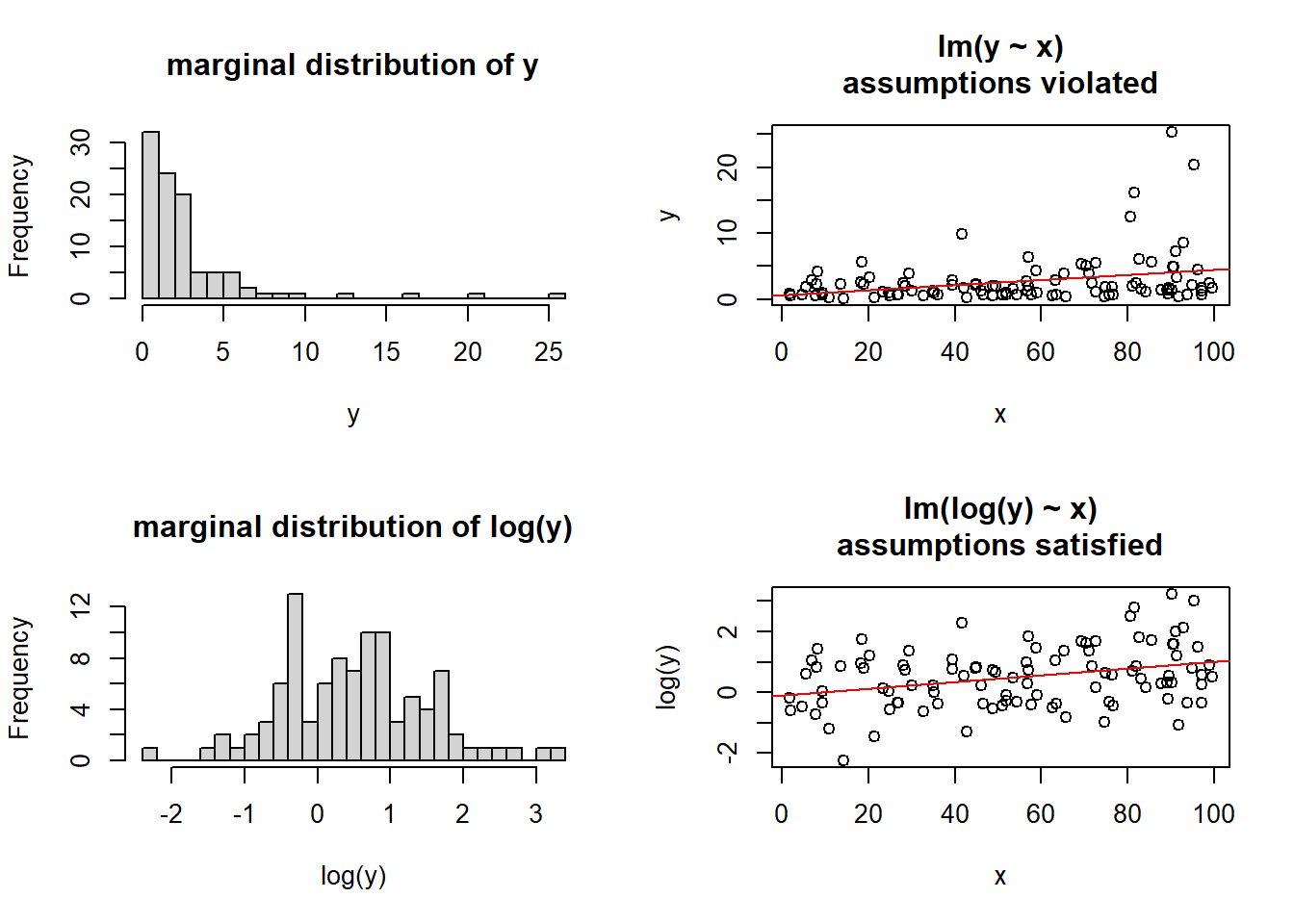
For the marginal distributions we will use density and boxplots, and for the bivariate associations a scatterplot.
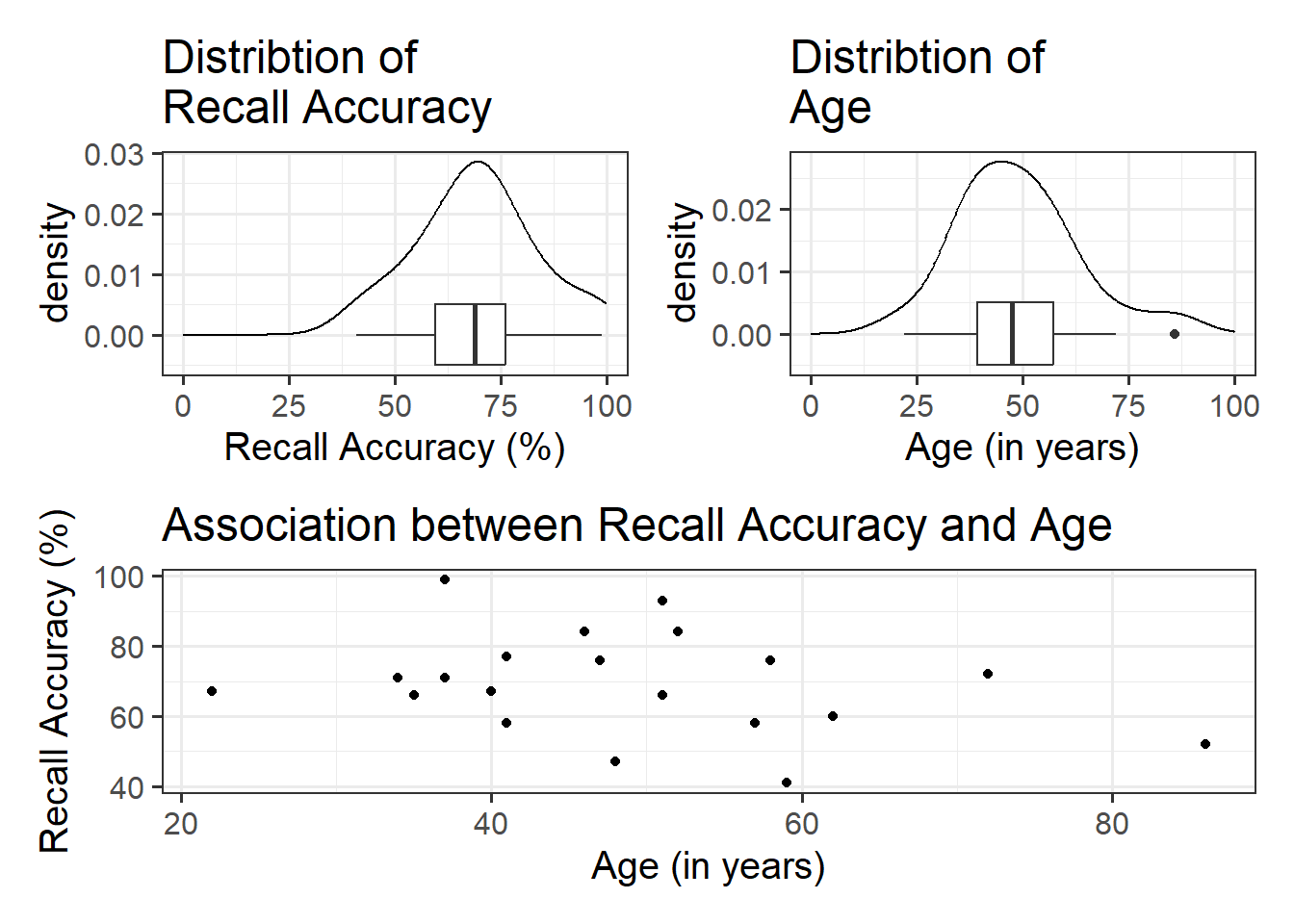
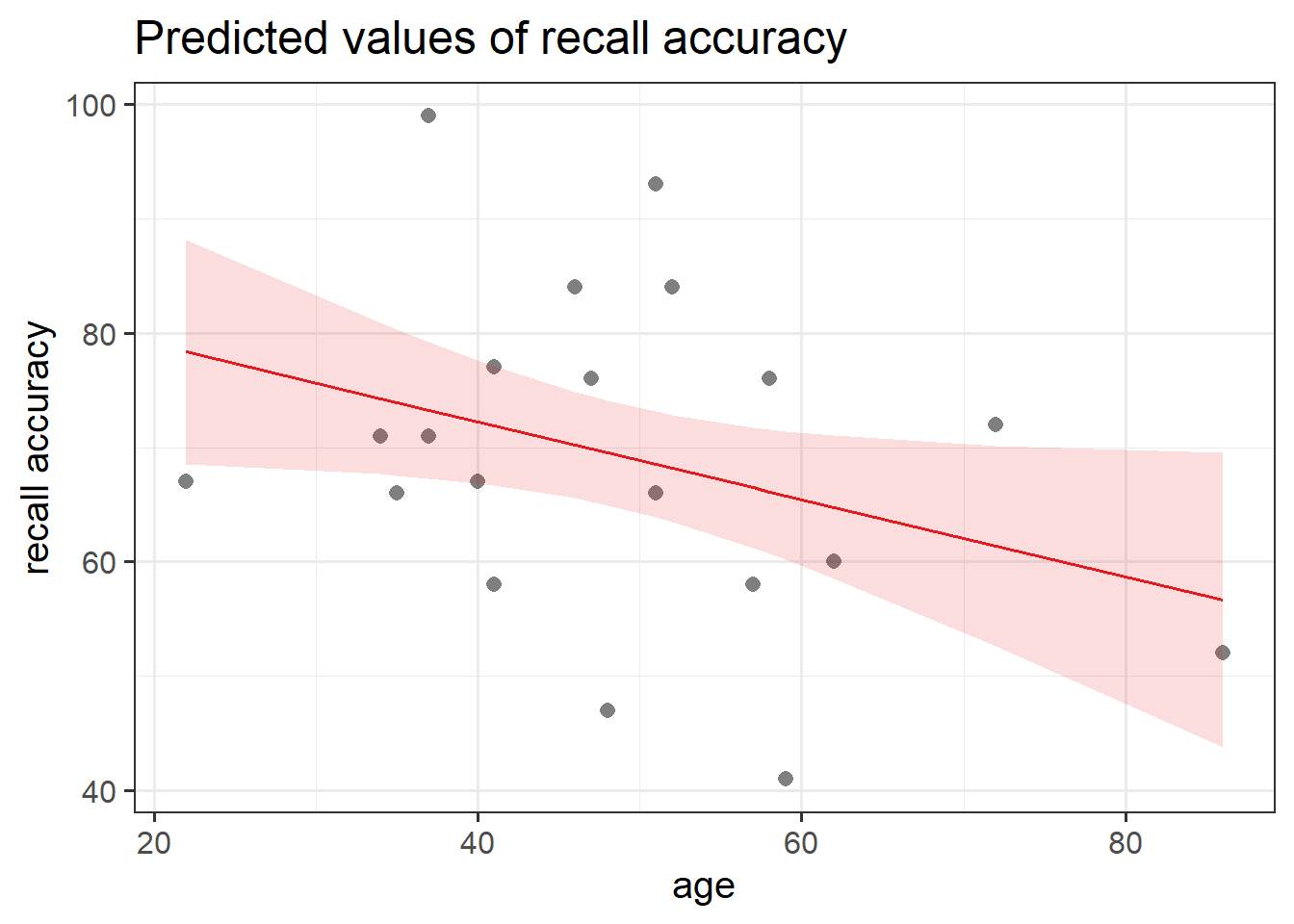
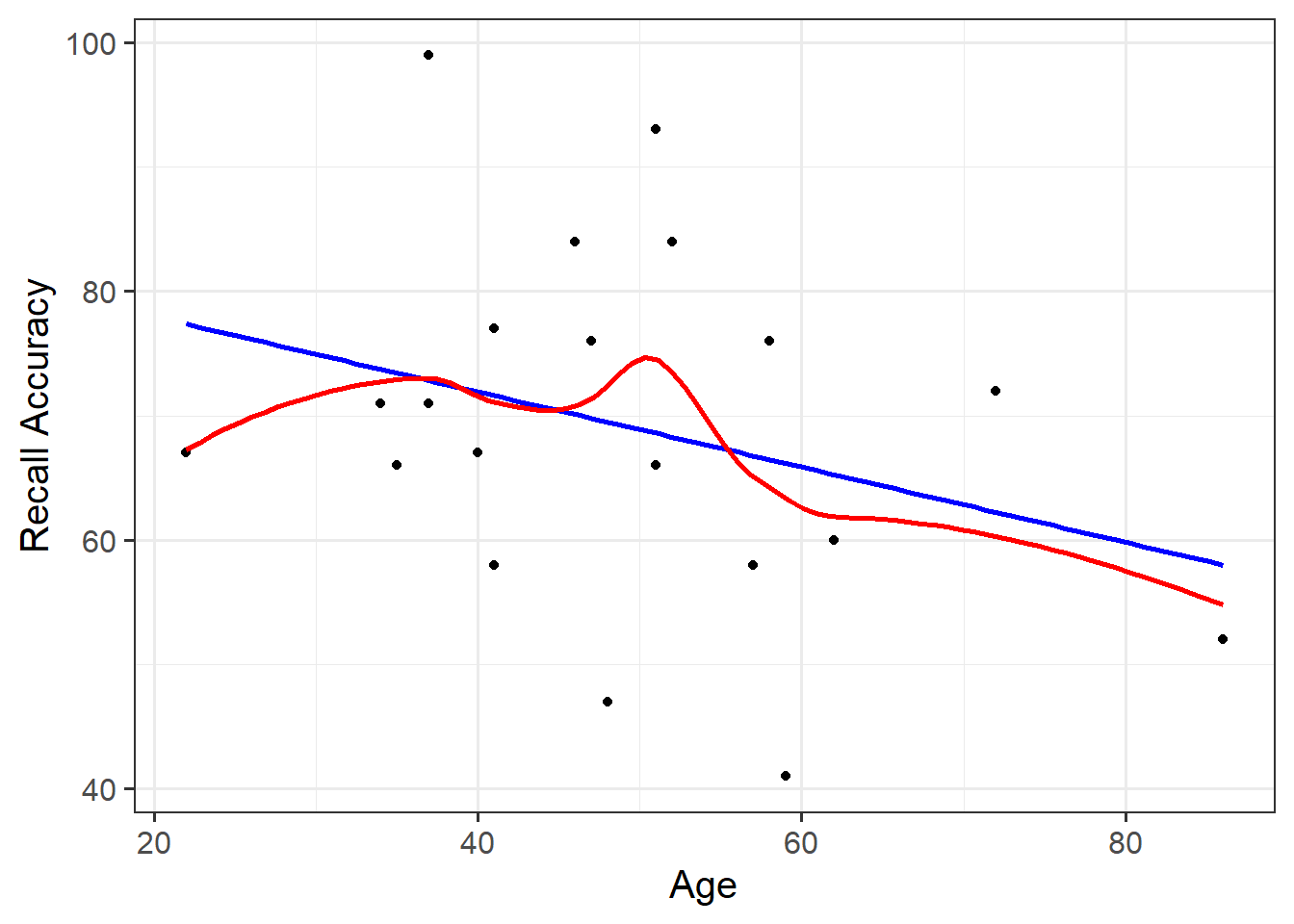
#save plots to individual objects in order to arrange plt1<-ggplot(data =recalldata, aes(x =recall_accuracy))+geom_density()+xlim(0, 100)+#specify x-axis to range from 0-100geom_boxplot(width =1/100)+labs(x ="Recall Accuracy (%)", title ="Distribtion of \nRecall Accuracy")plt2<-ggplot(data =recalldata, aes(x =age))+geom_density()+xlim(0, 100)+#specify x-axis to range from 0-100geom_boxplot(width =1/100)+labs(x ="Age (in years)", title ="Distribtion of \nAge")plt3<-ggplot(data =recalldata, aes(x =age, y =recall_accuracy))+geom_point()+labs(x ="Age (in years)", y ="Recall Accuracy (%)", title ="Association between Recall Accuracy and Age")#load patchwork package to arrange plotslibrary(patchwork)#arrange plots where there are two plots in to panel (plt1 + plt2), one on bottom (plt3)(plt1+plt2)/plt3
The marginal distribution of recall accuracy was unimodal with a negative skew with a mean of approximately 69.25. There was high variation in recall accuracy (SD = 14.53)
The marginal distribution of age was unimodal with a mean of approximately 48.8, where age ranged from 22 to 86
There appeared to be a weak negative association between recall accuracy and age, where older age was associated with lower recall accuracy
There is no association between recall accuracy and age.
\(H_1: \beta_1 \neq 0\)
There is an association between recall accuracy and age.
Model Building
To fit the model in R we use the lm() function. The simple linear model is assigned/stored in an object called recall_simp:
recall_simp<-lm(recall_accuracy~age, data =recalldata)
recall_simp
Call:
lm(formula = recall_accuracy ~ age, data = recalldata)
Coefficients:
(Intercept) age
84.0153 -0.3026
When we call the name of the fitted model, recall_simp, you can see the estimated regression coefficients \(\hat \beta_0\) and \(\hat \beta_1\). The line of best-fit is thus given by:1
The intercept, or predicted recall accuracy when age was 0.
An individual aged 0 years was expected to have a recall accuracy of \(84.02\).
Note: the intercept isn’t very useful here at all. It estimates the accuracy for a newborn (who wouldn’t be able to complete the task!).
\(\beta_1\) = age = -0.3
The estimated difference in recall accuracy for each additional year in age.
Every 1 additional year in age was associated with a non-significant \(-0.3\) percentage point decrease in recall accuracy \((p = .196)\). This suggested that age was not significantly associated with recall accuracy.
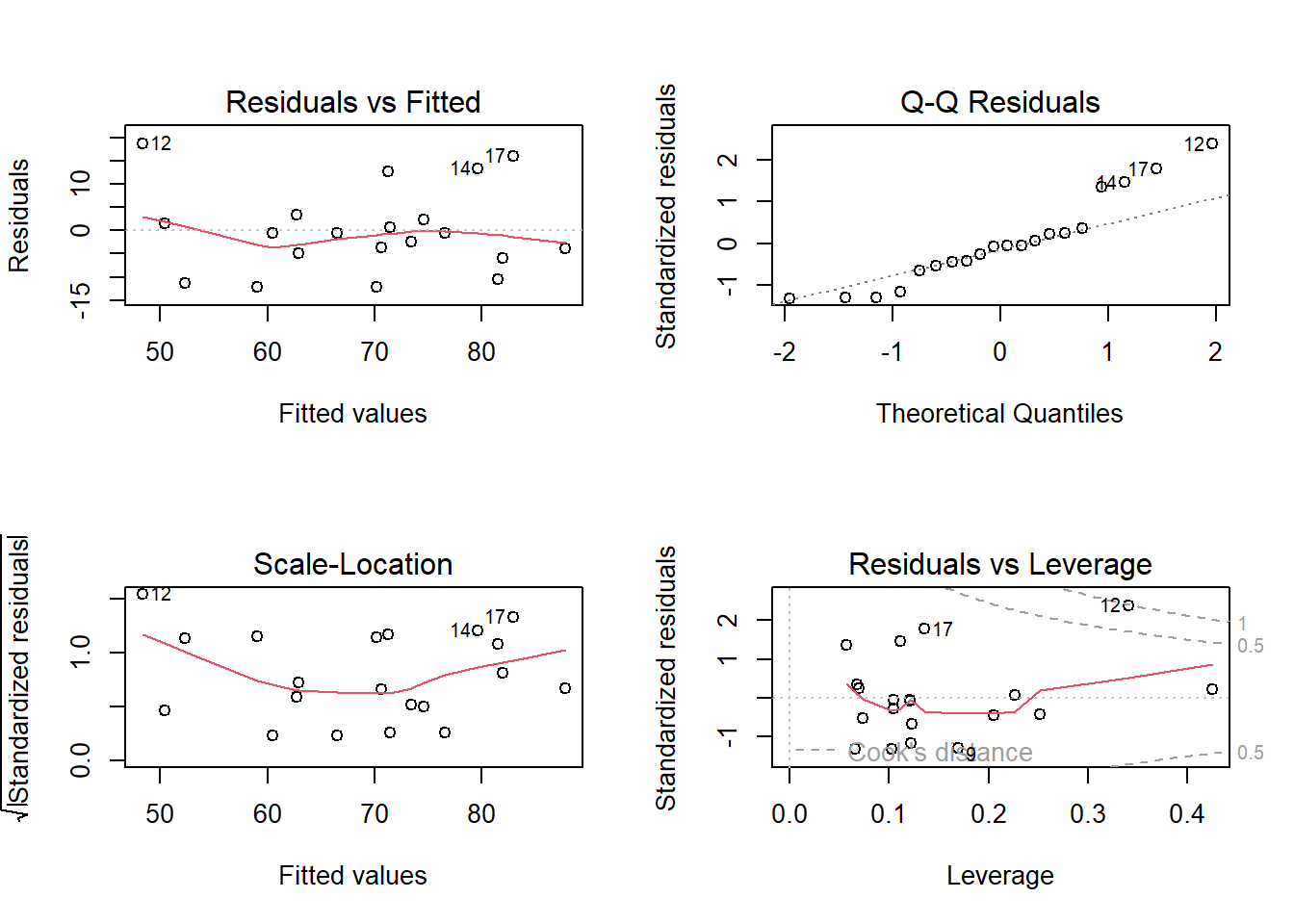
Model Visualisation
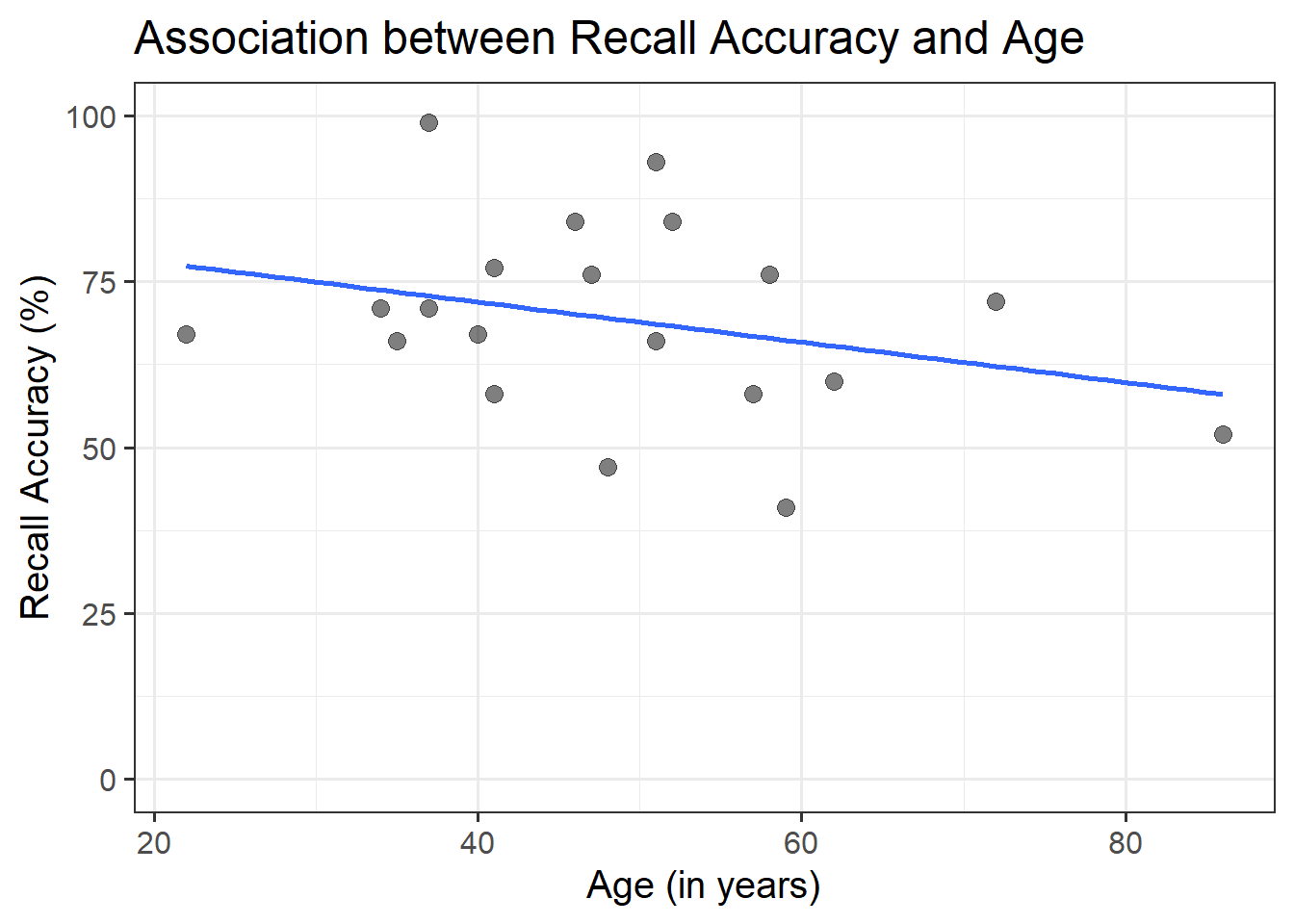
ggplot(recalldata, aes(x =age, y =recall_accuracy))+geom_point(size =3, alpha =0.5)+geom_smooth(method =lm, se =FALSE)+ylim(0,100)+labs(x ="Age (in years)", y ="Recall Accuracy (%)", title ="Association between Recall Accuracy and Age")
Figure 6: Association between Recall Accuracy and Age
The line that best fits the association between recall accuracy and age (see Figure 6) is only able to predict the average accuracy for a given value of age.
This is because there will be a distribution of recall accuracy at each value of age. The line will fit the trend/pattern in the values, but there will be individual-to-individual variability that we must accept around that average pattern.
Multiple Linear Regression Models
Description & Model Specification
Multiple linear regression involves looking at one continuous outcome (i.e., DV), with two or more independent variables (i.e., IVs).
A multiple linear regression model takes the following form:
Multiple and simple linear regression follow the same structure within the lm() function - the logic scales up to however many predictor variables we want to include in our model. You simply add (using the + sign) more independent variables. For example, if we wanted to build a multiple linear regression that included three independent variables, we could fit one of the following via the lm() function:
You’ll hear a lot of different ways that people explain multiple regression coefficients.
For the model \(y = \beta_0 + \beta_1 \cdot x_1 + \beta_2 \cdot x_2 + \epsilon\), the estimate \(\hat \beta_1\) will often be reported as:
“the increase in \(y\) for a one unit increase in \(x_1\) when…”
“holding the effect of \(x_2\) constant.”
“controlling for differences in \(x_2\).”
“partialling out the effects of \(x_2\).”
“holding \(x_2\) equal.”
“accounting for effects of \(x_2\).”
For models with 3+ predictors, just like building the model in R, the logic of the above simply extends.
For example “the increase in [outcome] for a one unit increase in [predictor] when…”
“holding [other predictors] constant.”
“accounting for [other predictors].”
“controlling for differences in [other predictors].”
“partialling out the effects of [other predictors].”
“holding [other predictors] equal.”
“accounting for effects of [other predictors].”
Example
Research Question
Is recall accuracy associated with recall confidence and age?
Overview
Imagine that you were tasked to investigate whether recall accuracy was associated with recall confidence and age. You have been provided with data from twenty participants who studied passages of text (c500 words long), and were tested a week later. The testing phase presented participants with 100 statements about the text. They had to answer whether each statement was true or false, as well as rate their confidence in each answer (on a sliding scale from 0 to 100). The dataset contains, for each participant, the percentage of items correctly answered, their age (in years), and their average confidence rating.
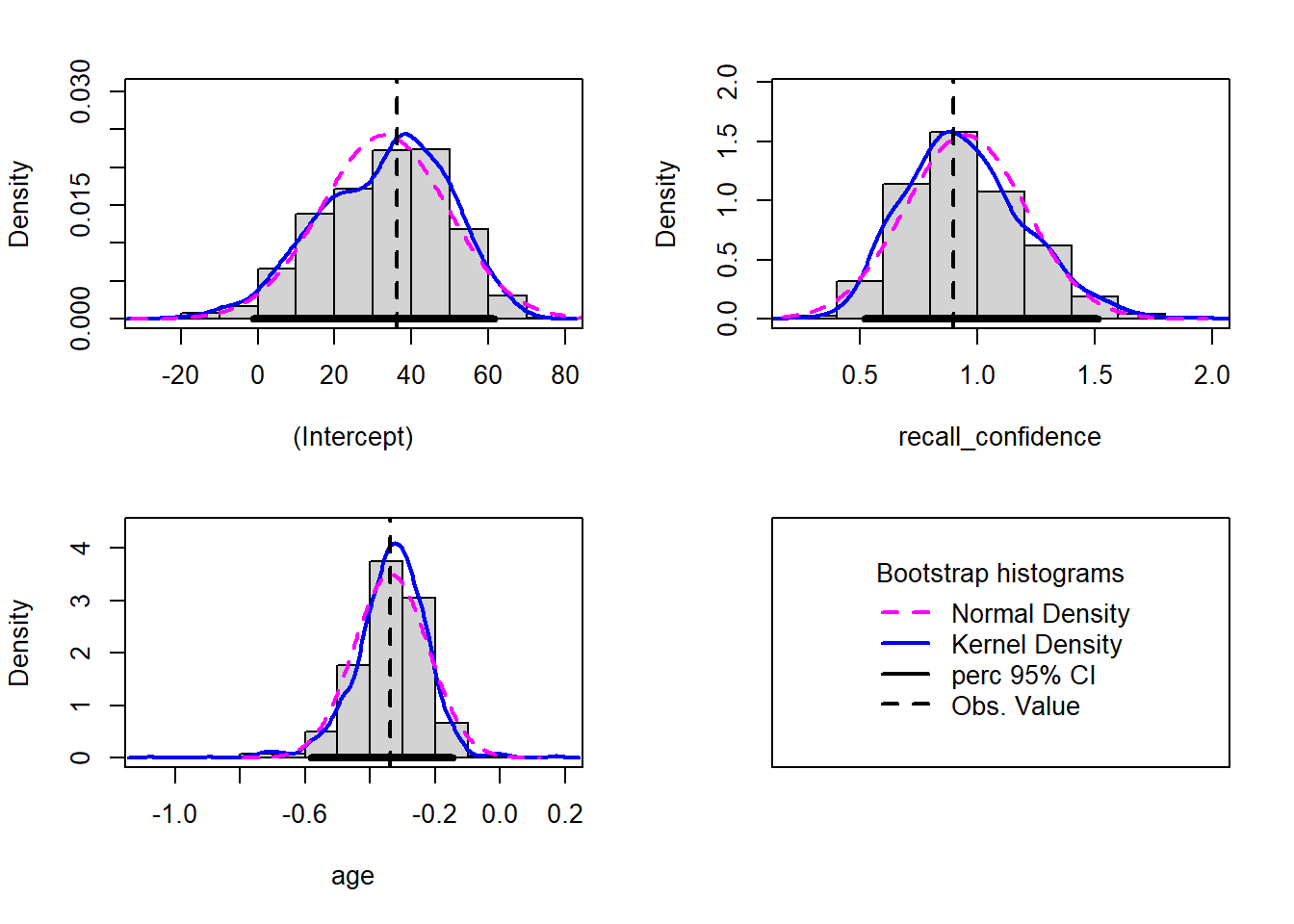
The intercept, or predicted recall accuracy when recall confidence was 0 and age was 0.
An individual aged 0 years with no recall confidence was expected to have a recall accuracy of \(36.16\).
Note: the intercept isn’t very useful here at all. It estimates the accuracy for a newborn (who wouldn’t be able to complete the task!).
\(\beta_1\) = recall_confidence = 0.9
The estimated difference in recall accuracy for each additional unit increase in confidence controlling for age.
Holding age constant, each 1 additional unit in recall confidence was associated with a significant \(0.9\) percentage point increase in recall accuracy \((p < .001)\).
\(\beta_2\) = age = -0.34
The estimated difference in recall accuracy for each additional year in age controlling for recall confidence.
Holding recall confidence constant, every 1 additional year in age was associated with a significant \(-0.34\) percentage point decrease in recall accuracy \((p = .041)\).
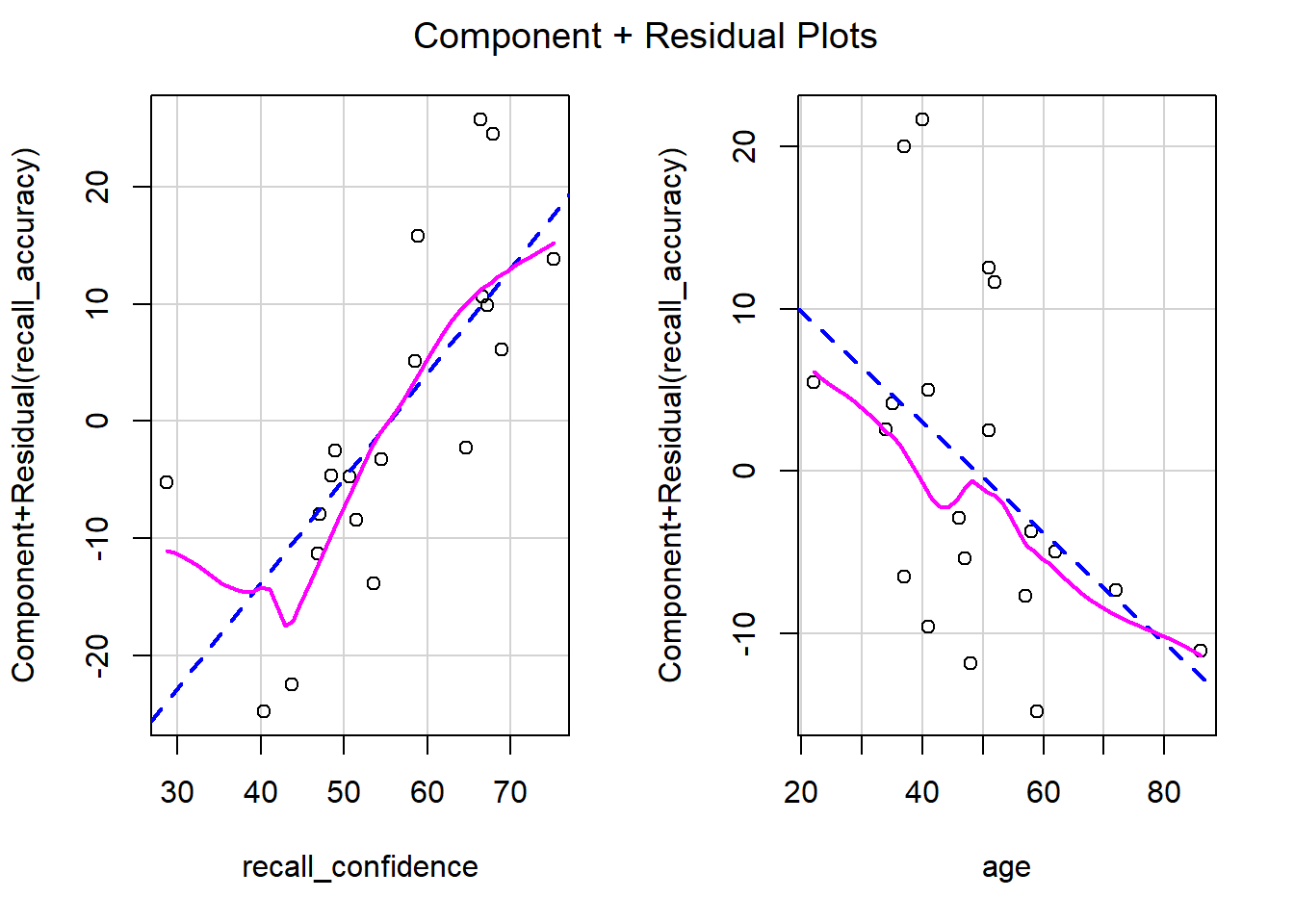
Model Visualisation
When we have 2+ predictors, we can’t just plot our data an add geom_smooth(method=lm), because that would give a visualisation of a linear model with just one predictor (whichever one is on the \(x\)-axis).
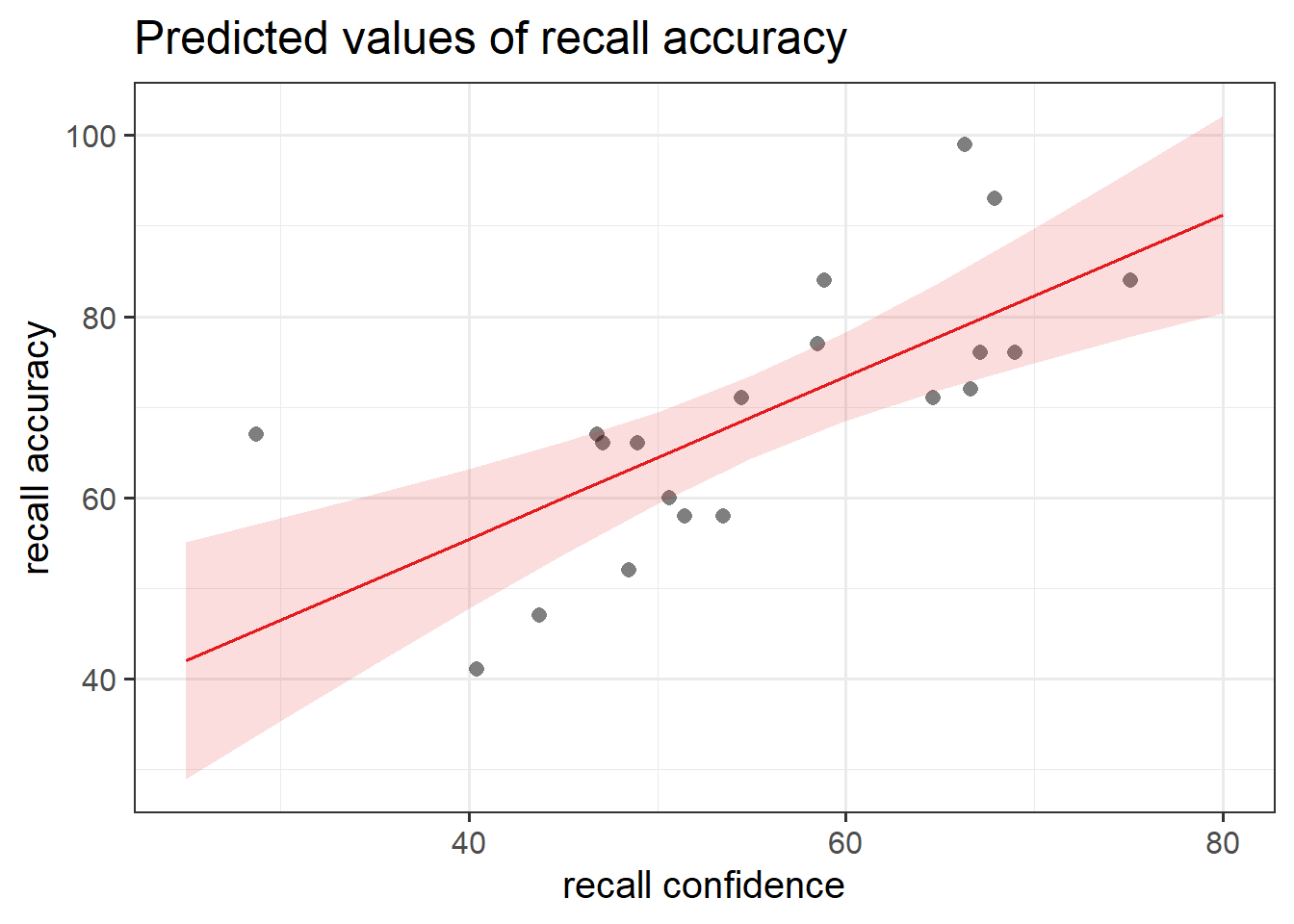
Instead, we can use the function plot_model() from sjPlot.
plot_model(recall_multi, type ="eff", terms ="recall_confidence", show.data =TRUE)plot_model(recall_multi, type ="eff", terms ="age", show.data =TRUE)
Figure 8: Association between Recall Accuracy, Recall Confidence, and Age
Figure 9: Association between Recall Accuracy, Recall Confidence, and Age
Numeric Outcomes & Categorical Predictors
Description & Model Specification
In both simple and multiple linear regression models, we examine the association between one continuous dependent variable (DV) using 1 (in the case of simple) or 2+ (in the case of multiple) predictor variables, which could include categorical predictors. When incorporating categorical variables, we use techniques such as dummy/treatment or effects/sum-to-zero coding to convert and represent these categorical predictors in a numerical format which is suitable for regression analysis.
The interpretation of the coefficients is very specific. Whereas we talked about coefficients being interpreted as “the change in \(y\) associated with a 1-unit increase in \(x\)”, for categorical explanatory variables, coefficients can be considered to examine differences in group means.
In R:
Multiple and simple linear regression models with categorical predictors follow the same structure within the lm() function. You simply add (using the + sign) more independent variables. For example, if we wanted to build a linear regression model that included one independent categorical variable which had three levels, we could fit one of the following via the lm() function:
Binary variables have two categories (more commonly referred to as levels), and these levels (e.g., Yes/No, Dog/Cat, Right/Left, Smoker/Non-Smoker) are simply entered in the model as a series of 0s and 1s. Numeric variables that represent categorical data are typically referred to as dummy variables.
Our coefficients are just the same as before. The intercept is where our predictor equals zero, and the slope is the change in our outcome variable associated with a 1-unit change in our predictor.
However, “zero” for this predictor variable now corresponds to a whole level. This is known as the “reference level”. Accordingly, the 1-unit change in our predictor (the move from “zero” to “one”) corresponds to the difference between the two levels.
When used as predictors in multiple regression models, binary variables behave much the same way. The coefficient will give us the estimated change in \(y\) when moving from one level to the other, whilst holding other predictors constant (for more info, see the Multiple Linear Regression Models - Interpretation of Coefficients flashcard).
Dummy vs Effects Coding
Possible side-constraints on the parameters are:
Name
Constraint
Meaning of \(\beta_0\)
In R
Sum to zero (Effects Coding)
\(\beta_1 + \beta_2 + \beta_3 = 0\)
\(\beta_0 = \mu\)
contr.sum
Reference group (Dummy Coding)
\(\beta_1 = 0\)
\(\beta_0 = \mu_1\)
contr.treatment
Dummy Coding
By default R uses the reference group constraint - i.e., dummy coding (sometimes called treatment contrast coding). If your factor has \(g\) levels, your regression model will have \(g-1\) dummy variables (R creates them for you, as we’ve seen in the examples above).
One level of the categorical variable is considered as the ‘baseline’, ‘reference level’, or ‘reference group’ - R automatically takes the first level alphabetically as the baseline (e.g., if you had a ‘pet’ variable with levels dog, hamster, and cat, then cat would be taken as the reference level).
When we use this approach, the intercept is the estimated \(y\) when all predictors (i.e., \(x\)’s) are zero. Because the reference level is kind of like “0” in our contrast matrix, this is part of the intercept estimate. We get out a coefficient for each subsequent level, which are the estimated differences from each level to the reference group.
Effects Coding
Effects coding (sometimes called sum-to-zero coding) is the next most commonly used in psychological research. These are a way of comparing each level to the overall (or grand) mean. This involves a bit of trickery that uses -1s and 1s rather than 0s and 1s, in order to make “0” be mid-way between all the levels - the average of the levels.
R automatically takes the last level alphabetically as level which is dropped (e.g., if you had a ‘pet’ variable with levels dog, hamster, and cat, then hamster would not be represented).
When we use this approach, the intercept is the estimated average \(y\) when averaged across all levels of the predictor variable. In other words, it is the estimated grand mean of \(y\). The coefficients represent the estimated difference for that level from the overall grand mean.
As a first step, it is a good idea to look at the structure of the dataset you are working with. For the purpose of this example, our dataset is called “tips” (you might recall this from DAPR1):
spc_tbl_ [157 × 7] (S3: spec_tbl_df/tbl_df/tbl/data.frame)
$ Bill : num [1:157] 23.7 36.1 32 17.4 15.4 ...
$ Tip : num [1:157] 10 7 5.01 3.61 3 2.5 3.44 2.42 3 2 ...
$ Credit: chr [1:157] "n" "n" "y" "y" ...
$ Guests: num [1:157] 2 3 2 2 2 2 2 2 2 2 ...
$ Day : chr [1:157] "f" "f" "f" "f" ...
$ Server: chr [1:157] "A" "B" "A" "B" ...
$ PctTip: num [1:157] 42.2 19.4 15.7 20.8 19.5 13.4 16 12.4 12.7 10.7 ...
- attr(*, "spec")=
.. cols(
.. Bill = col_double(),
.. Tip = col_double(),
.. Credit = col_character(),
.. Guests = col_double(),
.. Day = col_character(),
.. Server = col_character(),
.. PctTip = col_double()
.. )
- attr(*, "problems")=<externalptr>
From the output, we can see that Credit (whether guests paid with a credit card; n/y responses) was coded as a <chr> or character variable. If we wanted to set this as a factor so that R recognises it as a categorical variable, we can use on of the following:
We could also use the factor() function, and at the same time label factors appropriately to aid reader interpretation (it may not be immediately clear to some that n represents ‘No’ and y represents ‘Yes’). To do so, we list the all levels of Credit, and provide a new label corresponding to each level:
When you have a categorical variable coded as a factor, R will default to using alphabetical ordering. We could override this by making it a factor with an ordering to it’s levels (see the use of factor() and levels()). Functions like fct_relevel() might be handy too.
Example
Research Question
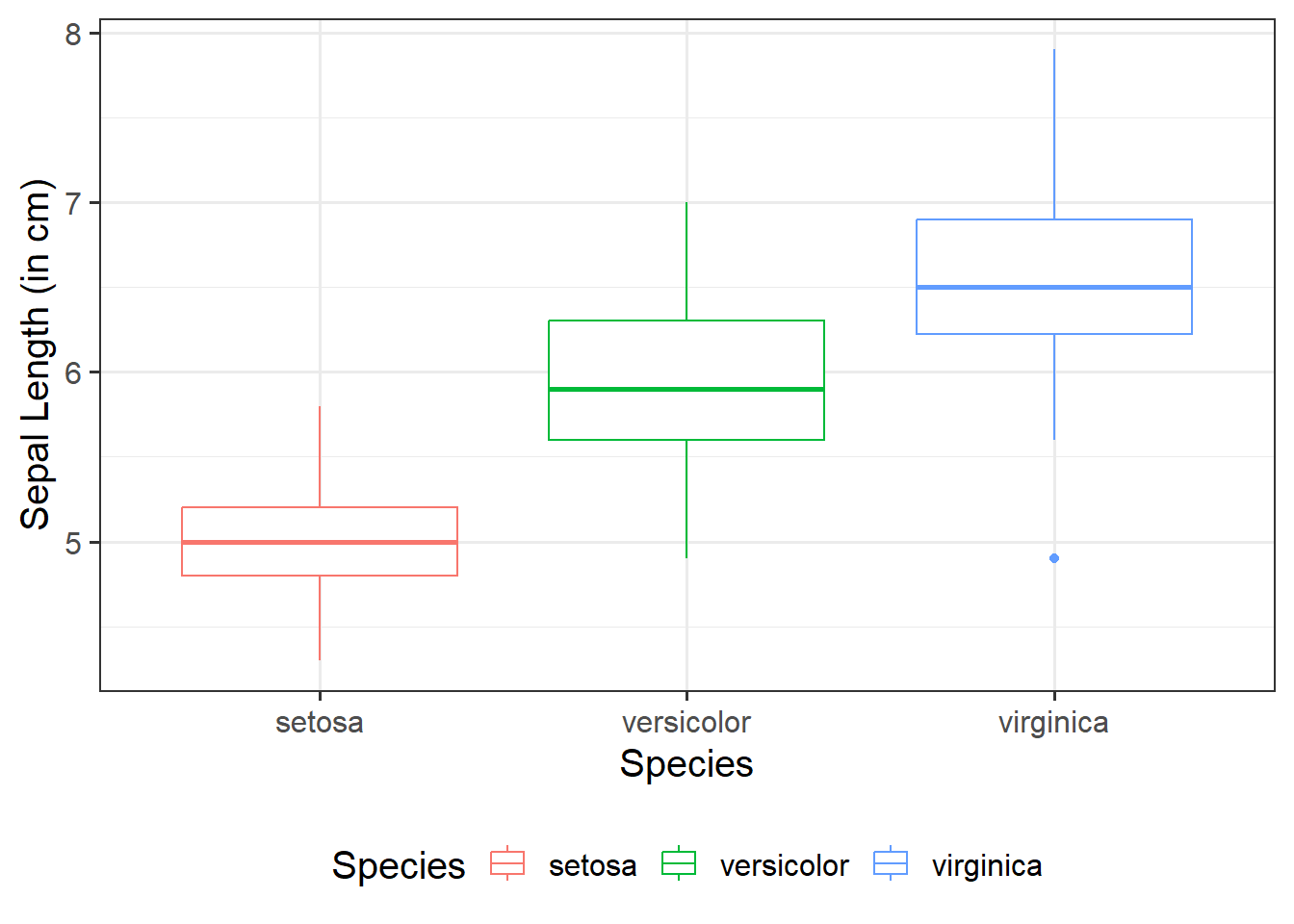
Does sepal length differ by species?
Overview
Imagine that you were tasked to investigate whether sepal length differs by species. You have been provided with the in-built dataset, iris, which contains information concerning the sepal length (in cm), sepal width (in cm), petal length (in cm), and petal width (in cm) from three different species of iris (setosa, versicolor, and virginica). There are measurements for 50 flowers from each of the iris species (i.e., total \(n\) = 150).
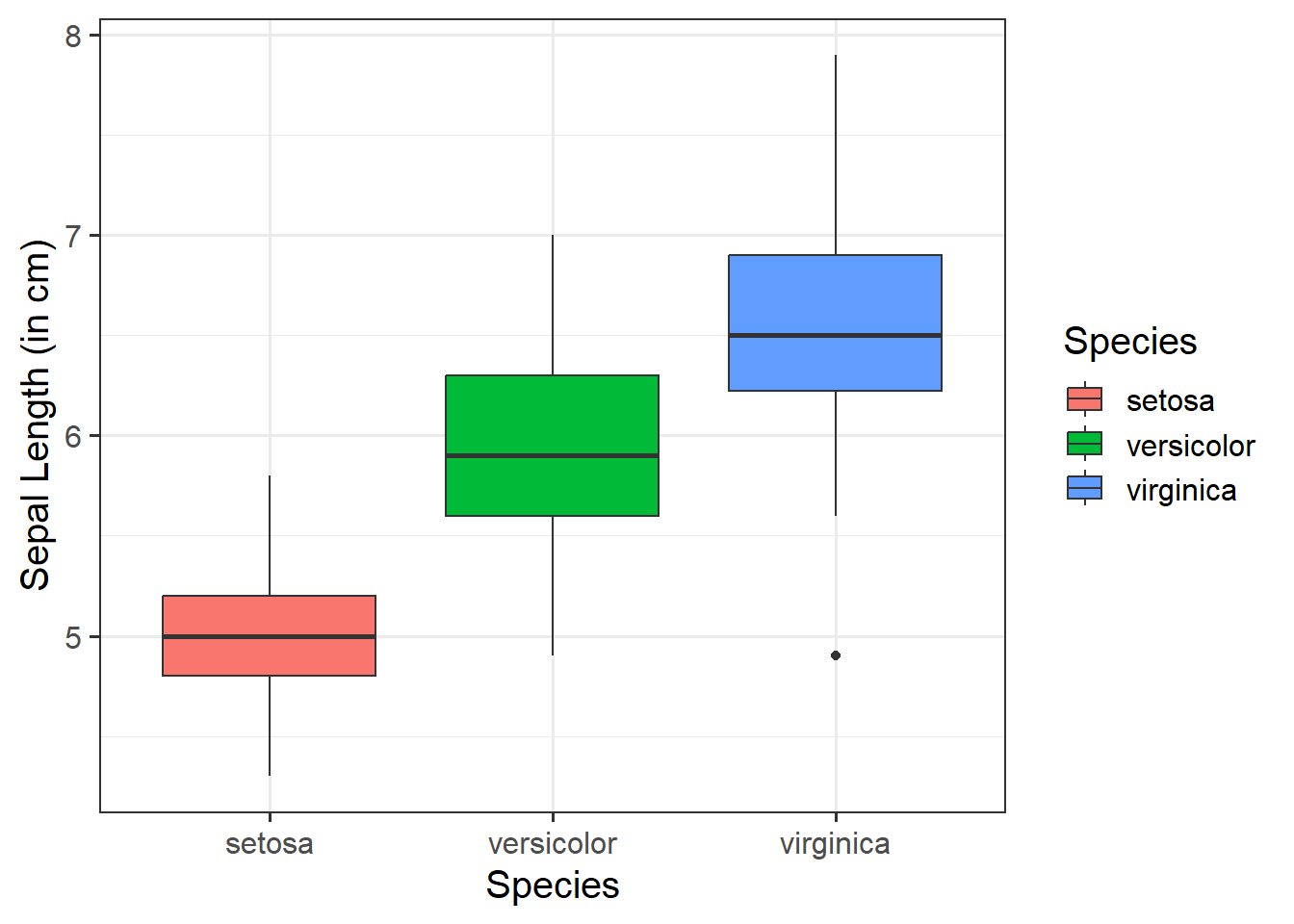
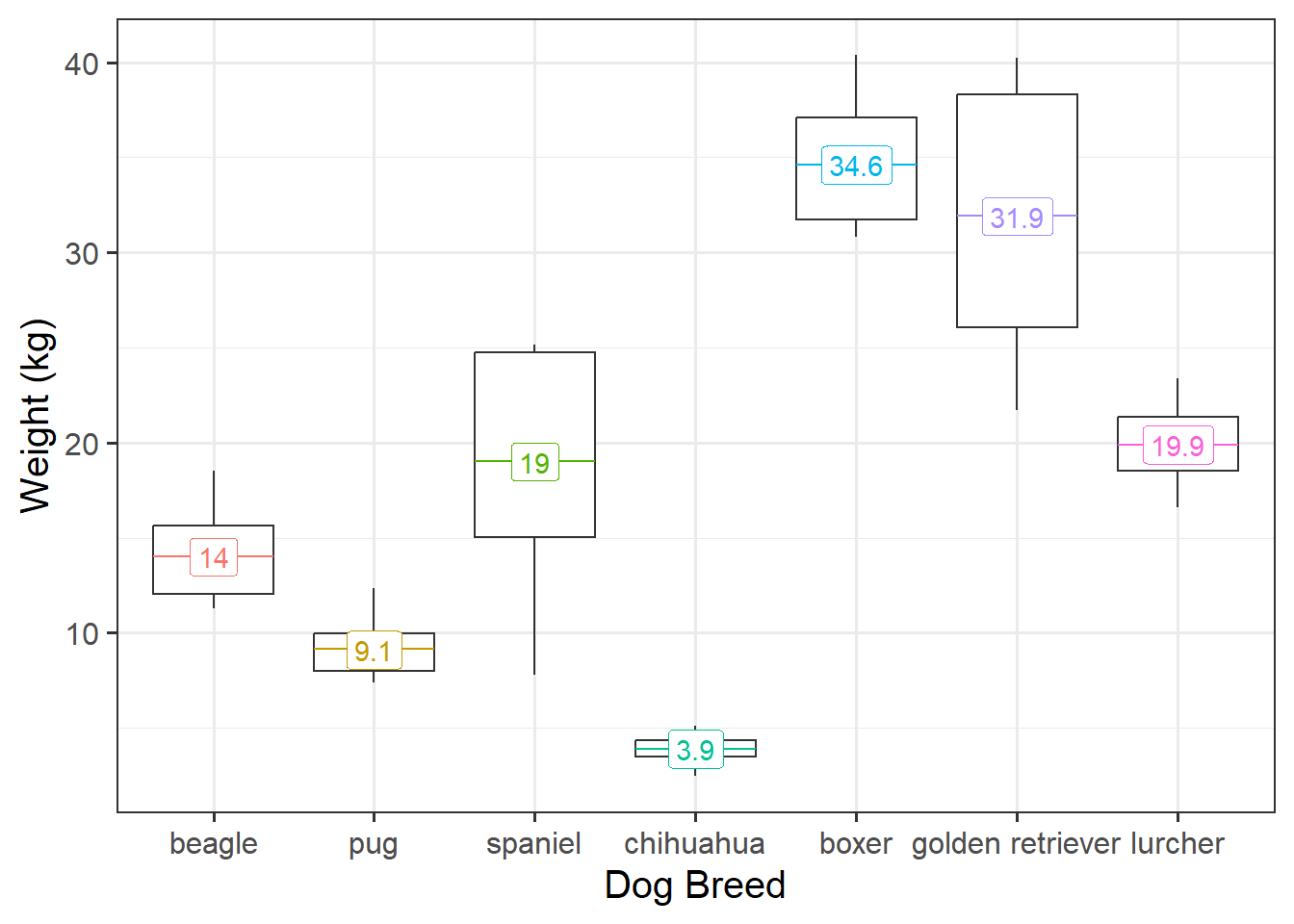
Visualise Data
ggplot(data =iris, aes(x =Species, y =Sepal.Length, fill =Species))+geom_boxplot()+labs(x ="Species", y ="Sepal Length (in cm)")
From the above, we can see that Species has 3 levels - “setosa”, “versicolor”, and “virginica”.
If we put these into a model, assuming R’s default ordering, we know that R will automatically apply dummy (or treatment coding, i.e., contrasts(iris$Species) <- "contr.treatment"), and “setosa” will be taken as our reference group:
#fit modelspec_model<-lm(Sepal.Length~Species, data =iris)summary(spec_model)
Call:
lm(formula = Sepal.Length ~ Species, data = iris)
Residuals:
Min 1Q Median 3Q Max
-1.6880 -0.3285 -0.0060 0.3120 1.3120
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 5.0060 0.0728 68.762 < 2e-16 ***
Speciesversicolor 0.9300 0.1030 9.033 8.77e-16 ***
Speciesvirginica 1.5820 0.1030 15.366 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.5148 on 147 degrees of freedom
Multiple R-squared: 0.6187, Adjusted R-squared: 0.6135
F-statistic: 119.3 on 2 and 147 DF, p-value: < 2.2e-16
First we need to tell R to apply effects (or sum-to-zero) coding and check the ordering of the levels:
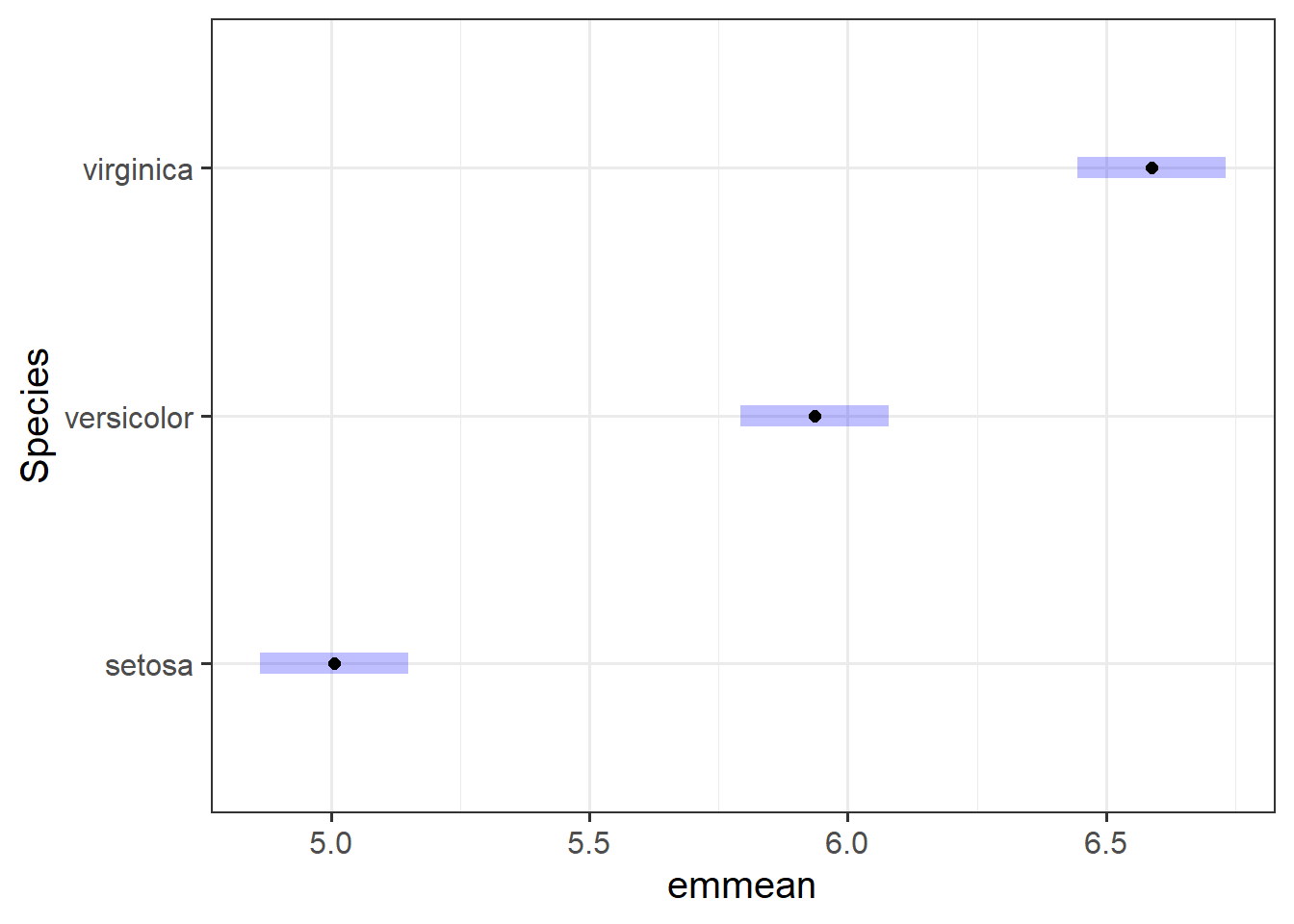
The estimate corresponding to (Intercept) contains \(\hat \beta_0 = \hat \mu_1 = 5.01\). The estimated average sepal length for the species setosa was approximately \(5.01~cm\).
The second estimate corresponds to Speciesversicolor and was \(\hat \beta_1 = 0.93\). The difference in mean sepal length between setosa and versicolor species was estimated to be \(0.93~cm\). Thus, \(\hat \mu_2 = 5.01 + 0.93 = 5.94\). We could say - the species iris versicolor had a sepal length of approximately \(5.94~cm\), and this was approximately \(0.93~cm\) longer than the iris setosa. This difference was statistically significant \((p < .001)\).
The third estimate corresponds to Speciesvirginica and was \(\hat \beta_2 = 1.58\). The difference in mean sepal length between setosa and virginica species was estimated to be \(1.58~cm\). Thus, \(\hat \mu_2 = 5.01 + 1.58 = 6.59\). We could say - the species iris virginica had a sepal length of approximately \(6.59~cm\), and this was approximately \(1.58~cm\) longer than the iris setosa. This difference was statistically significant \((p < .001)\).
The first estimate corresponding to (Intercept) contains \(\hat \beta_0 = \hat \mu = 5.84\). The estimated average sepal length across iris species was approximately \(5.84~cm\).
The second estimate corresponds to Species1 and was \(\hat \beta_1 = -0.84\). The difference in mean sepal length between setosa\((\hat \mu_1)\) and the grand mean \((\hat \mu_0)\) was estimated to be \(0.84~cm\). In other words, the iris species of setosa had a sepal length \(0.84~cm\) shorter than average, where its length was estimated to be \(5.84333 + (-0.83733) = 5~cm\). This difference in length was statistically significant \((p < .001)\).
The third estimate corresponds to Species2 and was \(\hat \beta_2 = 0.09\). The difference in mean sepal length between versicolor\((\hat \mu_2)\) and the grand mean \((\hat \mu_0)\) was estimated to be \(0.09~cm\). In other words, the iris species of versicolor had a sepal length \(0.09~cm\) longer than average, where its length was estimated to be \(5.84333 + 0.09267 = 5.94~cm\). This difference in length was not statistically significant \((p = .121)\).
The estimate for Species3, representing the difference of “virginica” to the grand mean is not shown by summary(). Because of the side-constraint, we know that \(\mu_3 = \beta_0 - (\beta_1 + \beta_2)\). The difference in sepal length between virginica and the grand mean was estimated to be \(-(-0.83733 + 0.09267) = 0.74466\). In other words, the virginica iris species had a sepal length \(0.74~cm\) longer than average, where its length was estimated to be \(5.84333 - (-0.83733 + 0.09267) = 6.59~cm\).
Model Visualisation
There are a couple of ways that we can visualise our model, eithe rusing the sjPlot or effects packages:
#fit modelspec_model2<-lm(Sepal.Length~Species, data =iris)summary(spec_model2)
Call:
lm(formula = Sepal.Length ~ Species, data = iris)
Residuals:
Min 1Q Median 3Q Max
-1.6880 -0.3285 -0.0060 0.3120 1.3120
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 5.84333 0.04203 139.020 <2e-16 ***
Species1 0.09267 0.05944 1.559 0.121
Species2 -0.83733 0.05944 -14.086 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.5148 on 147 degrees of freedom
Multiple R-squared: 0.6187, Adjusted R-squared: 0.6135
F-statistic: 119.3 on 2 and 147 DF, p-value: < 2.2e-16
General - Extracting Information
It is important to have a good grasp of how to understand and interpret the key components of your model summary() output, including model coefficients, standard errors, \(t\)-values, \(p\)-values, etc., and how these can be used in further calculations (such as confidence intervals). As well as knowing how to extract from R, it is necessary to understand how to compute some of these statistics by hand too.
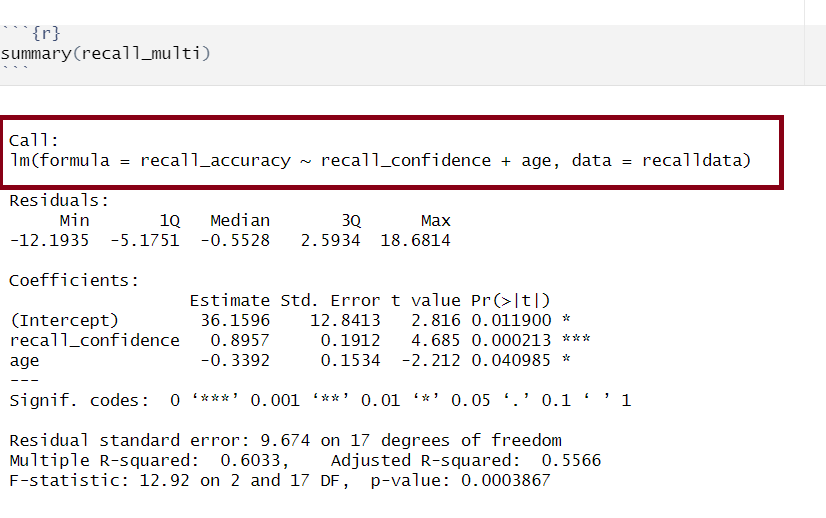
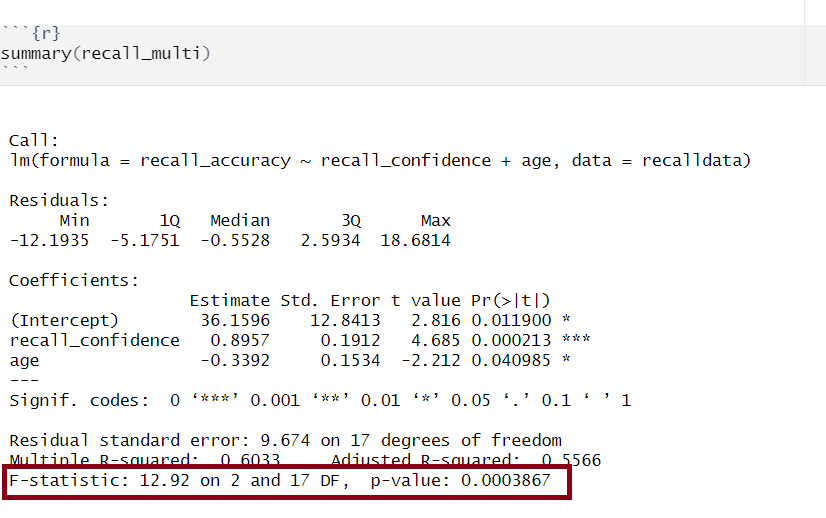
Model Call
Multiple regression output in R, model formula highlighted
The call section at the very top of the summary() output shows us the formula that was specified in R to fit the regression model.
In the above, we can see that recall accuracy is our DV, recall confidence and age were our two IVs, and our dataset was named recalldata.
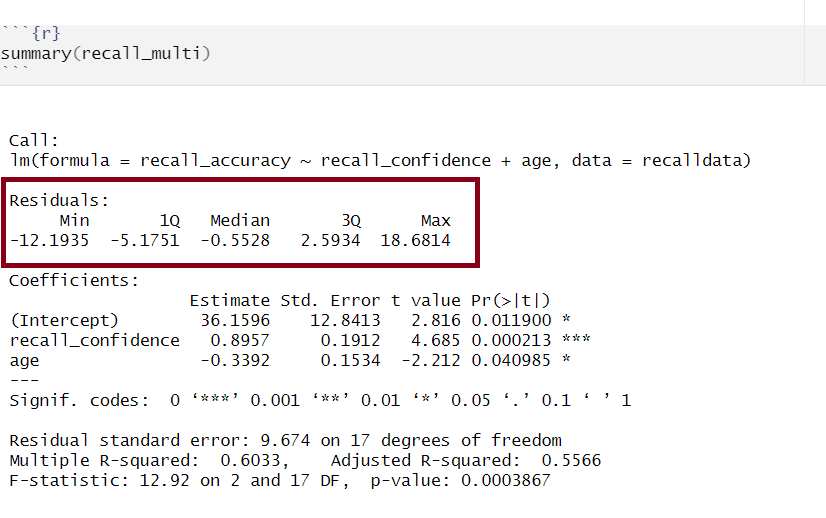
Residuals
Multiple regression output in R, residuals highlighted
Residuals are the difference between the observed values and model predicted values of the DV.
Ideally, for the model to be unbiased, we want our median value (the middle value of the residuals when ordered) to be around 0, as this would show that the errors are random fluctuations around the true line. When this is the case, we know that our model is doing a good job predicting values at the high and low ends of our dataset, and that our residuals were somewhat symmetrical.
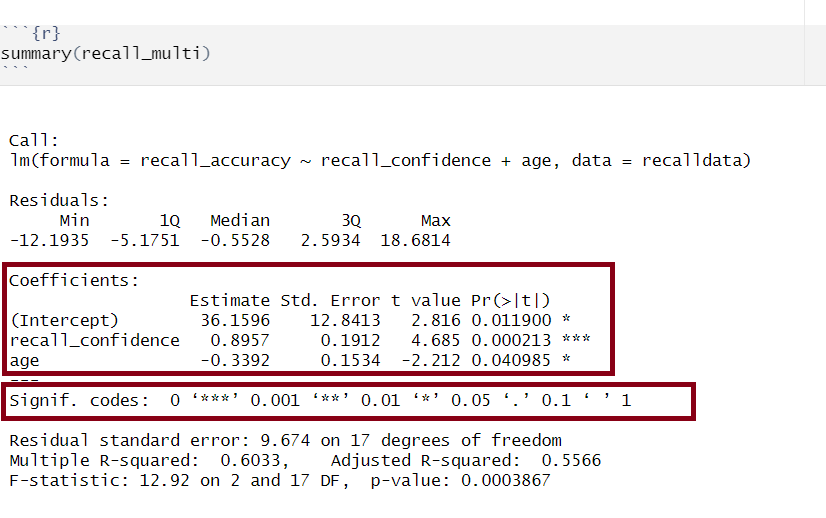
Model Coefficients
Multiple regression output in R, model coefficients highlighted
Let’s apply to a straightforward example to try by-hand. Suppose you have a simple linear regression model (i.e., with only one IV) where you have the following data points:
Observed \(x_i\)
Observed \(y_i\)
1
5
2
7
3
8
4
6
5
9
Step 1: Calculate mean of both \(x\) and \(y\)
\(\bar x = {\frac{1+2+3+4+5}{5}} = 3\)
\(\bar y = {\frac{5+7+8+6+9}{5}} = 7\)
Step 2: Calculate \(\beta_0\) and \(\beta_1\)
We need to calculate the slope first, as we need to know the value of \(\beta_1\) in order to calculate \(\beta_0\)
There are numerous equivalent ways to obtain the estimated regression coefficients — that is, \(\hat \beta_0\), \(\hat \beta_1\), …., \(\hat \beta_k\) — from the fitted model (for this below example, our fitted model has been named mdl):
mdl
mdl$coefficients
coef(mdl)
coefficients(mdl)
The standard error of the coefficient is an estimate of the standard deviation of the coefficient (i.e., how much uncertainty there is in our estimated coefficient).
The formula for the standard error of the slope is:
\[
\begin{align}
& SE(\hat \beta_j) = \sqrt{\frac{\text{SS}_\text{Residual}/(n-k-1)}{\sum(x_{ij} - \bar{x_{j}})^2(1-R_{xj}^2)}} \\
\\
& \text{Where}: \\
\\
& \text{SS}_\text{Residual} = \text{ residual sum of squares} \\
& n = \text{ sample size} \\
& k = \text{ number of predictors} \\
& x_{ij} = \text{ the observed value of a predictor (j) for an individual (i)} \\
& \bar{x_{j}} = \text{the mean of a predictor (j)} \\
& R_{xj}^2 = \text{the multiple correlation coefficient of the predictors} \\
\end{align}
\]
Let’s apply to a straightforward example. Suppose you have a simple linear regression model (i.e., with only one IV, which means that \(R_{xj}^2 = 0\) since there is only one predictor) and the following data points:
Observed \(x_i\)
Observed \(y_i\)
1
5
2
7
3
8
4
6
5
9
There are a number of steps you need to take to calculate by hand:
Calculate sum of the squared residuals
Calculate predicted values
Calculate residuals (i.e., the difference between the observed value (\(y_i\)) and the predicted value (\(\hat{y}_i\)) for each observation)
Square the residuals
Calculate the Sum of Squared Residuals
Calculate the sum of squared deviations of the (\(x\)) values from their mean
Use values from 1 & 2 to calculate \(SE(\hat \beta_j)\)
Step 1.1: Calculate predicted values
Using \(\hat{y}_i = \beta_0 + \beta_1 \cdot x_i\) and our model coefficients \(\beta_0 = 4.9\) and \(\beta_1 = 0.7\):
Step 2. Calculate the sum of squared deviations of the (\(x\)) values from their mean
The mean of \(x\) can be calculated as: \(\bar x = {\frac{1+2+3+4+5}{5}} = 3\). Using this, we can then calculate the sum of squared deviations of \(x\):
If you wanted to obtain just the standard error for each estimated regression coefficient, you could do the following (for this below example, our fitted model has been named mdl):
summary(mdl)$coefficients[,2]
The t-statistic is the \(\beta\) coefficient divided by the standard error:
\[
t = \frac{\hat \beta_j - 0}{SE(\hat \beta_j)}
\]
which follows a \(t\)-distribution with \(n-k-1\) degrees of freedom (where \(k\) = number of predictors and \(n\) = sample size).
With this, we can test the the null hypothesis \(H_0: \beta_j = 0\).
Generally speaking, you want your model coefficients to have large \(t\)-statistics as this would indicate that the standard error was small in comparison to the coefficient. The larger our \(t\)-statistic, the more confident we can be that the coefficient is not 0.
How to calculate \(t = \frac{\hat \beta_j - 0}{SE(\hat \beta_j)}\)
(Intercept) recall_confidence age
2.815890 4.684654 -2.211515
From our \(t\)-value, we can compute our \(p\)-value. The \(p\)-value help us to understand whether our coefficient(s) are statistically significant (i.e., that the coefficient is statistically different from 0). The \(p\)-value of each estimate indicates the probability of observing a \(t\)-value at least as extreme as, or more extreme than, the one calculated from the sample data when assuming the null hypothesis to be true.
In Psychology, a \(p\)-value < .05 is usually used to make statements regarding statistical significance (it is important that you always state your \(\alpha\) level to help your reader understand any statements regarding statistical significance).
The number of asterisks marks corresponds with the significance of the coefficient (see the ‘Signif. codes’ legend just under the coefficients section).
In R
If you wanted to obtain just the \(p\)-values for each estimated regression coefficient, you could do the following (for this below example, our fitted model has been named mdl):
summary(mdl)$coefficients[,4]
Confidence Intervals
Using the estimate and standard error of a given \(\beta\) coefficient, we can create confidence intervals to estimate a plausible range of values for the true population parameter. Recall the formula for obtaining a confidence interval for the population slope is:
\[
\hat \beta_j \pm t^* \cdot SE(\hat \beta_j)
\]
where \(t^*\) denotes the critical value chosen from \(t\)-distribution with \(n-k-1\) degrees of freedom (where \(k\) = number of predictors and \(n\) = sample size) for a desired \(\alpha\) level of confidence.
How to calculate \(\hat \beta_j \pm t^* \cdot SE(\hat \beta_j)\)
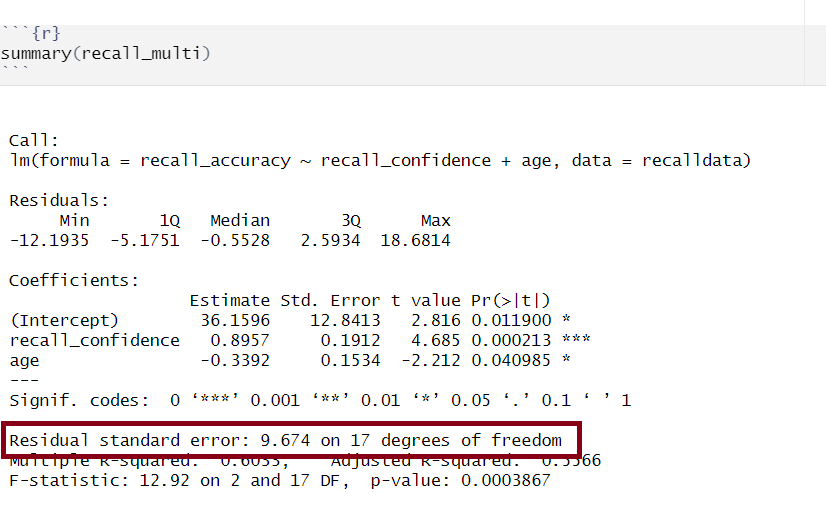
Multiple regression output in R, model standard deviation of the errors highlighted
The standard deviation of the errors, denoted by \(\sigma\), is an important quantity that our model estimates. It represents how much individual data points tend to deviate above and below the regression line - in other words, it tells us how well the model fits the data.
A small \(\sigma\) indicates that the points hug the line closely and we should expect fairly accurate predictions, while a large \(\sigma\) suggests that, even if we estimate the line perfectly, we can expect individual values to deviate from it by substantial amounts.
The estimated standard deviation of the errors is denoted \(\hat \sigma\), and is estimated by essentially averaging squared residuals (giving the variance) and taking the square-root:
There are a couple of equivalent ways to obtain the estimated standard deviation of the errors — that is, \(\hat \sigma\) — from the fitted model (for this example, our fitted model has been named mdl):
sigma(mdl)
summary(mdl)
Manual Contrasts
Dummy and effects coding allow us to test the significance of the difference between means of groups and some other mean (either reference group or grand mean respectively). However, in some cases, we may want to test more specific hypotheses that require us to test the difference between particular combinations of groups. In such cases, we can use manual contrasts.
Rules & General Process
There are some rules that we need to follow when using manual contrasts:
Rule 1: Weights are -1 \(\geq\) x \(\leq\) 1
Rule 2: The group(s) in one chunk are given negative weights, the group(s) in the other get positive weights
Rule 3: The sum of the weights of the comparison must be 0
Rule 4: If a group is not involved in the comparison, weight is 0
Rule 5: For a given comparison, weights assigned to group(s) are equal to 1 divided by the number of groups in that chunk.
Rule 6: Restrict yourself to running \(k\) - 1 comparisons (where \(k\) = number of groups)
Rule 7: Each contrast can only compare 2 chunks of variance
Rule 8: Once a group singled out, it can not enter other contrasts
Following the above, we can implement as follows:
Step 1: ‘Chunk’ together the groups that the research question is interested in comparing
Step 2: Assign a 0 to any group(s) that aren’t in one of the chunks from Step 1
Step 3: Assign a plus sign to every group in Chunk 1, and a minus sign to every group in Chunk 2
Step 4: Count the plus signs and minus signs
Step 5: To figure out the actual values for each cell, start with 1 and –1 Divide 1 by \(n_{plus}\), and divide –1 by \(n_{minus}\)
Step 6: In the coding matrix, replace the plus signs with the positive coding value from Step 5, and replace the minus signs with the negative coding value from Step 5. And that’s it done!
Additive Models
Steps In R
After specifying our hypotheses, to test our contrasts, we can use the emmeans package and follow the below structure:
Step 2: Use the emmeans() function to obtain the estimated means of each group. You can visualise these by using plot() on the obtained estimated means of the groups
Step 3: Check the order of your levels via levels()
Step 4: Define the contrast by specifying the weights following the rules outlined above (as well as paying attention to the ordering of the levels)
Step 5: Test the pre-specified group contrast(s) via contrast()
After completing these steps, the last task would be to interpret the results of the contrast analysis in the context of the hypothesis.
Example
Research Question
Does the sepal length of an iris grown in Western states (i.e., iris setosa) differ from the sepal length of an Iris grown in Eastern states (i.e., iris versicolor and virginica)?
Specify Hypotheses
Based on the research question, we are asking whether there is a difference between the average sepal length of iris setosa (Western states), and the combined average sepal length of iris versicolor and iris virginica (Eastern state)s. To assess this for the Eastern states, we need to compute the average of the mean sepal lengths of the two species, iris versicolor and iris virginica.
With this in mind, we could specify our hypotheses as:
contrast estimate SE df lower.CL upper.CL
Western State Iris - Eastern State Iris 1.26 0.0892 147 1.08 1.43
Confidence level used: 0.95
# Bonus Step: Run inferential test and return CIs in one commandsummary(seplength_comp_test, infer =TRUE)
contrast estimate SE df lower.CL upper.CL
Western State Iris - Eastern State Iris 1.26 0.0892 147 1.08 1.43
t.ratio p.value
14.086 <.0001
Confidence level used: 0.95
Results Interpretation
Important to write up our findings in the context of the hypothesis / research question:
We performed a test against \(H_0: \mu_\text{Setosa} = \frac{1}{2} (\mu_\text{Versicolor} + \mu_\text{Virginica}) = 0\). At the 5% significance level, there was evidence that iris sepal length significantly differed between Western and Eastern states in the US \((t(147) = 14.09, p < .001, \text{two-sided})\), and this difference was estimated to be \(1.26~cm\). We are 95% confident that an iris grown in an Eastern state, on average, would be between \(1.08~cm\) and \(1.43~cm\) longer than those grown in a Western state \((CI_{95}[1.08, 1.43])\).
Model Predicted Values & Residuals
Model predicted values are the estimates generated by a regression model for the dependent variable based on the independent variable(s), whilst residuals are the differences between these predicted values and the actual observed values (in turn indicating the accuracy of the model’s predictions).
Predicted Values
Model predicted values (\(\hat y_i\)) for sample data
We can get out the model predicted values for \(y\), the “y hats” (\(\hat y\)), for the data in the sample using various functions:
predict(<fitted model>)
fitted(<fitted model>)
fitted.values(<fitted model>)
mdl$fitted.values
For example, this will give us the estimated recall accuracy (point on our regression line) for each observed value of age for each of our 20 participants.
Model predicted values for other (unobserved) data
To compute the model-predicted values for unobserved data (i.e., data not contained in the sample), we can use the following function:
predict(<fitted model>, newdata = <dataframe>)
For this example, we first need to remember that the model predicts recall_accuracy using the independent variable age. Hence, if we want predictions for new (unobserved) data, we first need to create a tibble with a column called age containing the age of individuals for which we want the prediction, and store this as a dataframe.
#Create dataframe 'newdata' containing the age values of 19, 32, and 99newdata<-tibble(age =c(19,32,99))newdata
# A tibble: 3 × 1
age
<dbl>
1 19
2 32
3 99
Then we take newdata and add a new column called accuracy_hat, computed as the prediction from the fitted recall_simp using the newdata above:
# A tibble: 3 × 2
age accuracy_hat
<dbl> <dbl>
1 19 78.3
2 32 74.3
3 99 54.1
Residuals
The residuals (\(\hat \epsilon_i\)) represent the deviations between the actual responses and the predicted responses and can be obtained either as
mdl$residuals
resid(mdl)
residuals(mdl)
computing them as the difference between the response (\(y_i\)) and the predicted response (\(\hat y_i\))
Predicted Values - Example
Lets estimate (or predict) recall accuracy of two individuals with the following ages (a) 18, and (b) 118. There are a few ways we can do this, but first, let’s recall our fitted model:
We can see that both approaches (manually substituting values into the regression equation or using the predict() function) both give us the same values (slightly different due to rounding).
But, be careful to not go too far off the range of the available data (I don’t know many 118 year olds, do you?). If you do, you will extrapolate. This is very dangerous…
Source: Randall Munroe, xkcd.com
Data Transformations
There are many transformations we can do to a continuous variable, but the most common ones are centering and scaling. These transformations can help to aid interpretability of our statistical models.
Centering
Centering simply means moving the entire distribution to be centered on some new value. We achieve this by subtracting our desired center from each value of a variable.
A common option is to mean center (i.e. to subtract the mean from each value). This makes our new values all relative to the mean. We can center a variable on other things, such as the minimum or maximum value of the scale we are using, or some judiciously chosen value of interest.
model<-lm(scale(DV, scale =FALSE)~scale(IV, scale =FALSE), data =data_name)
Scaling
Scaling changes the units of the variable, and we do this by dividing the observations by some value. E.g., moving from “36 months” to “3 years” involves multiplying (scaling) the value by 1/12.
The most common transformation that involves scaling is called standardisation.
Standardisation
This involves subtracting the mean from each individual observation (i.e., calculating individual deviations) and then dividing by the standard deviation. So standardisation centers on the sample mean and scales by the sample standard deviation.
Recall that a standardized variable has mean of 0 and standard deviation of 1. If both\(x\) and \(y\) are standardised, our model coefficients (\(\beta\)’s) are standardised too.
When we standardise variables in a regression model, it means we can talk about all our coefficients in terms of “standard deviation units”. To the extent that it is possible to do so, this puts our coefficients on scales of the similar magnitude, making qualitative comparisons between the sizes of effects a little more easy.
We tend to refer to coefficients using standardised variables as (unsurprisingly), “standardised coefficients”
There are two main ways that people construct standardised coefficients. One of which standardises just the predictor, and the other of which standardises both predictor and outcome:
predictor
outcome
in lm
coefficient
interpretation
standardised
raw
y ~ scale(x)
\(\beta = b \cdot s_x\)
“difference in Y for a 1 SD increase in X”
standardised
standardised
scale(y) ~ scale(x)
\(\beta = b \cdot \frac{s_x}{s_y}\)
“difference in SD of Y for a 1 SD increase in X”
Model Fit
Linear Models
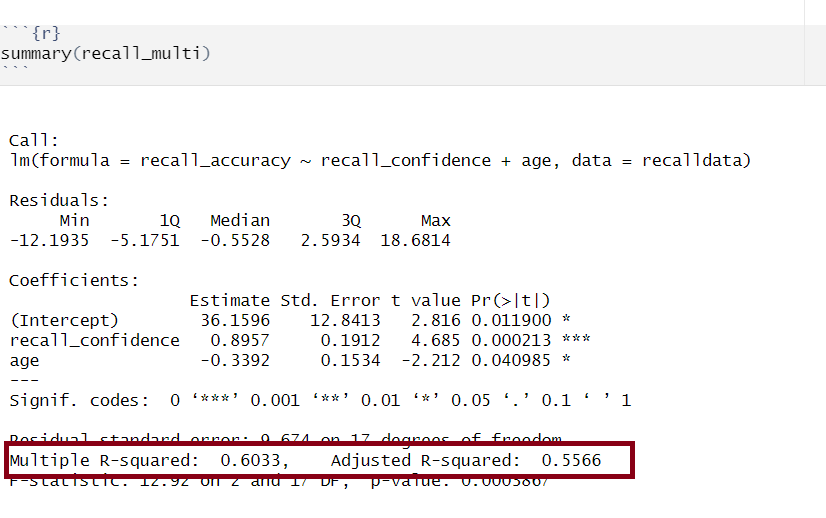
Assessing model fit involves examining metrics like the sum of squares to measure variability explained by the model, the \(F\)-ratio to evaluate the overall significance of the model by comparing explained variance to unexplained variance, and \(R\)-squared / Adjusted \(R\)-squared to quantify the proportion of variance in the dependent variable explained by the independent variable(s).
Sums of Squares
To quantify and assess a model’s utility in explaining variance in an outcome variable, we can split the total variability of that outcome variable into two terms: the variability explained by the model plus the variability left unexplained in the residuals.
The sum of squares measures the deviation or variation of data points away from the mean (i.e., how spread out are the numbers in a given dataset). We are trying to find the equation/function that best fits our data by varying the least from our data points.
Total Sum of Squares
Formula:
\[
\text{SS}_\text{Total} = \sum_{i=1}^{n}(y_i - \bar{y})^2
\] Can also be derived from:
Squared distance of each data point from the mean of \(y\).
Description:
How much variation there is in the DV.
Example:
Let’s apply to a straightforward example to try by-hand. Suppose you have a simple linear regression model (i.e., with only one IV) where you have the following data points:
Observed \(x_i\)
Observed \(y_i\)
1
5
2
7
3
8
4
6
5
9
Steps:
Calculate the mean of \(y\) (\(\bar y\))
Calculate for each observation \(y_i\) - \(\bar y\)
Square each of the obtained \(y_i\) - \(\bar y\) values
Sum squared values
Step 1: Calculate the mean of \(y_i\)
\(\bar y = {\frac{5+7+8+6+9}{5}} = 7\)
Step 2 & 3: Calculate for each observation \(y_i\) - \(\bar y\) & square values
Squared distance of each point from the predicted value.
Description:
How much of the variation in the DV the model did not explain - a measure that captures the unexplained variation in your regression model. Lower residual sum of squares suggests that your model fits the data well, and higher suggests that the model poorly explains the data (in other words, the lower the value, the better the regression model). If the value was zero here, it would suggest the model fits perfectly with no error.
Example:
Let’s apply to a straightforward example to try by-hand. Suppose you have a simple linear regression model (i.e., with only one IV) where you have the following data points:
Observed \(x_i\)
Observed \(y_i\)
1
5
2
7
3
8
4
6
5
9
Steps:
Calculate predicted values (\(\hat{y}_i\))
Calculate residuals (i.e., the difference between the observed value (\(y_i\)) and the predicted value (\(\hat{y}_i\)) for each observation)
Square the residuals
Sum squared values
Step 1: Calculate predicted values
Using \(\hat{y}_i = \beta_0 + \beta_1 \cdot x_i\) and our model coefficients \(\beta_0 = 4.9\) and \(\beta_1 = 0.7\):
The deviance of the predicted scores from the mean of \(y\).
Description:
How much of the variation in the DV your model explained - like a measure that captures how well the regression line fits your data.
Example:
Let’s apply to a straightforward example to try by-hand. Suppose you have a simple linear regression model (i.e., with only one IV) where you have the following data points:
Observed \(x_i\)
Observed \(y_i\)
1
5
2
7
3
8
4
6
5
9
Steps:
Calculate mean of \(y\) (\(\bar y\))
Calculate predicted values (\(\hat{y}_i\))
Calculate for each observation \(\hat{y}_i - \bar y\)
Squaring each of the obtained \(\hat{y}_i - \bar y\) values
Sum squared values
Step 1: Calculate the mean of \(y_i\)
\(\bar y = {\frac{5+7+8+6+9}{5}} = 7\)
Step 2: Calculate predicted values
Using \(\hat{y}_i = \beta_0 + \beta_1 \cdot x_i\) and our model coefficients \(\beta_0 = 4.9\) and \(\beta_1 = 0.7\):
Observed (\(x_i\))
Observed (\(y_i\))
Predicted (\(\hat{y}_i\))
1
5
\(4.9 + (0.7*1) = 5.6\)
2
7
\(4.9 + (0.7*2) = 6.3\)
3
8
\(4.9 + (0.7*3) = 7\)
4
6
\(4.9 + (0.7*4) = 7.7\)
5
9
\(4.9 + (0.7*5) = 8.4\)
Step 3 & 4: Calculate for each observation \(\hat{y}_i\) - \(\bar y\) & square values
We can perform a test to investigate if a model is ‘useful’ — that is, a test to see if our explanatory variable explains more variance in our outcome than we would expect by just some random chance variable.
With one predictor, the \(F\)-statistic is used to test the null hypothesis that the regression slope for that predictor is zero:
\[
H_0: \text{the model is ineffective, }b_1 = 0 \\
\]\[
H_1 : \text{the model is effective, }b_1 \neq 0 \\
\]
In multiple regression, the logic is the same, but we are now testing against the null hypothesis that all regression slopes are zero. Our test is framed in terms of the following hypotheses:
\[
H_0: \text{the model is ineffective, }b_1,...., b_k = 0 \\
\]
\[
H_1 : \text{the model is effective, }b_1,...., b_k \neq 0 \\
\]
The relevant test-statistic is the \(F\)-statistic, which uses “Mean Squares” (these are Sums of Squares divided by the relevant degrees of freedom). We then compare that against (you guessed it) an \(F\)-distribution! \(F\)-distributions vary according to two parameters, which are both degrees of freedom.
\[
\begin{align}
& \text{Where:} \\
& df_{model} = k \\
& df_{residual} = n-k-1 \\
& n = \text{sample size} \\
& k = \text{number of explanatory variables} \\
\end{align}
\]
Description:
To test the significance of an overall model, we can conduct an \(F\)-test. The \(F\)-test compares your model to a model containing zero predictor variables (i.e., the intercept only model), and tests whether your added predictor variables significantly improved the model.
It is called the \(F\)-ratio because it is the ratio of the how much of the variation is explained by the model (per parameter) versus how much of the variation is unexplained (per remaining degrees of freedom).
The \(F\)-test involves testing the statistical significance of the \(F\)-ratio.
Q: What does the \(F\)-ratio test? A: The null hypothesis that all regression slopes in a model are zero (i.e., explain no variance in your outcome/DV). The alternative hypothesis is that at least one of the slopes is not zero.
The \(F\)-ratio you see at the bottom of summary(model) is actually a comparison between two models: your model (with some explanatory variables in predicting \(y\)) and the null model.
In regression, the null model can be thought of as the model in which all explanatory variables have zero regression coefficients. It is also referred to as the intercept-only model, because if all predictor variable coefficients are zero, then we are only estimating \(y\) via an intercept (which will be the mean - \(\bar y\)).
Interpretation:
Alongside viewing the \(F\)-ratio, you can see the results from testing the null hypothesis that all of the coefficients are \(0\) (the alternative hypothesis is that at least one coefficient is \(\neq 0\). Under the null hypothesis that all coefficients = 0, the ratio of explained:unexplained variance should be approximately 1)
If your model predictors do explain some variance, the \(F\)-ratio will be significant, and you would reject the null, as this would suggest that your predictor variables included in your model improved the model fit (in comparison to the intercept only model).
Points to note:
The larger your \(F\)-ratio, the better your model
The \(F\)-ratio will be close to 1 when the null is true (i.e., that all slopes are zero)
The linear model with recall confidence and age explained a significant amount of variance in recall accuracy beyond what we would expect by chance \(F(2, 17) = 12.92, p < .001\).
R-squared and Adjusted R-squared
Overview:
\(R^2\) represents the proportion of variance in \(Y\) that is explained by the model predictor variables.
Formula:
The \(R^2\) coefficient is defined as the proportion of the total variability in the outcome variable which is explained by our model:
\[
\begin{align}
& \text{Where:} \\
& n = \text{sample size} \\
& k = \text{number of explanatory variables} \\
\end{align}
\]
When to report Multiple \(R^2\) vs. Adjusted \(R^2\):
The Multiple \(R^2\) value should be reported for a simple linear regression model (i.e., one predictor).
Unlike \(R^2\), Adjusted-\(R^2\) does not necessarily increase with the addition of more explanatory variables, by the inclusion of a penalty according to the number of explanatory variables in the model. Since Adjusted-\(R^2\) is adjusted for the number of predictors in the model, this should be used when there are 2 or more predictors in the model. As a side note, the Adjusted-\(R^2\) should always be less than or equal to \(R^2\).
How to calculate Multiple \(R^2\) & Adjusted \(R^2\)
Together, recall confidence and age explained approximately 55.66% of the variance in recall accuracy.
Model Comparisons
Linear Models
One useful thing we might want to do is compare our models with and without some predictor(s).There are numerous ways we can do this, but the method chosen depends on the models and underlying data:
Nested vs Non-Nested Models
Nested Models
Consider that you have two regression models where Model 1 contains a subset of the predictors contained in the other Model 2 and is fitted to the same data. More simply, Model 2 contains all of the predictors included in Model 1, plus additional predictor(s). This means that Model 1 is nested within Model 2, or that Model 1 is a submodel of Model 2. These two terms, at least in this setting, are interchangeable - it might be easier to think of Model 1 as your null and Model 2 as your alternative.
Non-Nested Models
Consider that you have two regression models where Model 1 contains different variables to those contained in Model 2, where both models are fitted to the same data. More simply, Model 1 and Model 2 contain unique variables that are not shared. This means that Model 1 and Model 2 are not nested.
Incremental F-test
If (and only if) two models are nested, can we compare them using an incremental F-test.
This is a formal test of whether the additional predictors provide a better fitting model.
Formally this is the test of:
\(H_0:\) coefficients for the added/omitted variables are all zero.
\(H_1:\) at least one of the added/omitted variables has a coefficient that is not zero.
The \(F\)-ratio for comparing the residual sums of squares between two models can be calculated as:
\[
F_{(df_R-df_F),~df_F} = \frac{(SSR_R-SSR_F)/(df_R-df_F)}{SSR_F / df_F} \\
\quad \\
\]\[
\begin{align}
& \text{Where:} \\
\\
& SSR_R = \text{residual sums of squares for the restricted model} \\
& SSR_F = \text{residual sums of squares for the full model} \\
& df_R = \text{residual degrees of freedom from the restricted model} \\
& df_F = \text{residual degrees of freedom from the full model} \\
\end{align}
\]
In R
We can conduct an incremental \(F\)-test to compare two models by fitting both models using lm(), and passing them to the anova() function:
If we wanted to, for example, compare a model with just one predictor, \(x1\), to a model with 2 predictors: \(x1\), and \(x2\), we can assess the extent to which the variable \(x2\) improves model fit:
model1<-lm(y~x1, data =data_name)model2<-lm(y~x1+x2, data =data_name)anova(model1, model2)
For example:
Model Comparisons using Incremental F-test
Example Interpretation
Recall confidence explained a significant amount of variance in recall accuracy beyond age \((F(1, 17) = 21.95, p < .001)\).
AIC & BIC
AIC (Akaike Information Criterion) and BIC (Bayesian Information Criterion) combine information about the sample size, the number of model parameters, and the residual sums of squares (\(SS_{residual}\)). Models do not need to be nested to be compared via AIC and BIC, but they need to have been fit to the same dataset.
\[
\begin{align}
& SS_{residual} = \text{sum of squares residuals} \\
& n = \text{sample size} \\
& k = \text{number of explanatory variables} \\
& \text{ln} = \text{natural log function}
\end{align}
\]
For both of these fit indices, lower values are better, and both include a penalty for the number of predictors in the model (although BIC’s penalty is harsher).
So how do we determine whether there is a statistical difference between two models? To evaluate our model comparisons, we need to look at the difference (\(\Delta\)) between the two values:
AIC: There are no specific thresholds to suggest how big a difference in two models is needed to conclude that one is substantively better than the other
BIC: Using the following \(\Delta BIC\) cutoffs (Raftery, 1995):
Value
Interpretation of Difference between Models
\(\Delta < 2\)
No evidence
\(2 > \Delta < 6\)
Positive evidence
\(6 > \Delta < 10\)
Strong evidence
\(\Delta > 10\)
Very strong evidence
In R
We can calculate AIC and BIC by using the AIC() and BIC() functions respectively:
Based on both AIC and BIC, the model predicting recall accuracy that included both recall confidence and age was better fitting \((\text{AIC} = 152.28; \text{BIC} = 156.27)\) than the model with age alone \((\text{AIC} = 166.86; \text{BIC} = 169.85)\).
Model Assumptions
Linear Models
Linear models rely on numerous underlying assumptions about the data. These assumptions ensure that the association between variables is appropriately captured, and that inferences drawn from the model are accurate and valid. Model diagnostics can help further assess whether these assumptions hold. When these assumptions are violated, there are numerous techniques that can be employed, such as through data transformations or using robust alternatives, to ensure reliable model interpretations.
Linearity
Simple Linear Regression
In simple linear regression with only one explanatory variable, we could assess linearity through a simple scatterplot of the outcome variable against the explanatory. This would allow us to check if the errors have a mean of zero. If this assumption was met, the residuals would appear to be randomly scattered around zero.
The rationale for this is that, once you remove from the data the linear trend, what’s left over in the residuals should not have any trend, i.e. have a mean of zero.
Multiple Regression
In multiple regression, however, it becomes more necessary to rely on diagnostic plots of the model residuals. This is because we need to know whether the relations are linear between the outcome and each predictor after accounting for the other predictors in the model.
In order to assess this, we use partial-residual plots (also known as ‘component-residual plots’). This is a plot with each explanatory variable \(x_j\) on the x-axis, and partial residuals on the y-axis***.
Partial residuals for a predictor \(x_j\) are calculated as: \[
\hat \epsilon + \hat \beta_j x_j
\]
#specify modelrecall_simp<-lm(recall_accuracy~age, data =recalldata)#create plotggplot(recalldata, aes(x =age, y =recall_accuracy))+geom_point()+geom_smooth(method ="lm", se =FALSE, colour ="blue")+#fit straight line to datageom_smooth(method ="loess", se =FALSE, colour ="red")+#fit loess line to datalabs(x ="Age", y ="Recall Accuracy")
Interpretation Guidance
The loess line should closely follow the data.
We can create these plots for all predictors in the model by using the crPlots() function from the car package:
#specify modelrecall_mdl<-lm(recall_accuracy~recall_confidence+age, data =recalldata)#create plotscrPlots(recall_mdl)
Interpretation Guidance
You are looking for the pink line to follow a linear trend line (i.e., follow the blue line). In other words, the loess line should closely follow the linear line.
Independence (of errors)
The ‘independence of errors’ assumption is the condition that the errors do not have some underlying relationship which is causing them to influence one another.
There are many sources of possible dependence, and often these are issues of study design. For example, we may have groups of observations in our data which we would expect to be related (e.g., multiple trials from the same participant). Our modelling strategy would need to take this into account.
Testing for the independence of errors can be pretty difficult, unless you know the potential source of correlation between cases (more on this in DAPR3!).
Normality (of errors)
The normality assumption is the condition that the errors \(\epsilon\) are normally distributed in the population.
We can visually assess this condition through histograms, density plots, and quantile-quantile plots (QQplots) of our residuals \(\hat \epsilon\).
Remember that departures from a linear trend in QQ plots indicate a lack of normality.
Equal Variances (Homoscedasticity)
The equal variances assumption is that the error variance \(\sigma^2\) is constant across values of the predictor(s) \(x_1, \dots, x_k\), and across values of the fitted values \(\hat y\). This sometimes gets termed “Constant” vs “Non-constant” variance. This is presented visually in Figure 10 and Figure 11.
Figure 10: Non-constant variance for numeric and categorical X
Figure 11: Constant variance for numeric and categorical X
In R
We can create plots of the Pearson residuals against the predicted values \(\hat y\) and against the predictors \(x_1\), … \(x_k\) by using the residualPlots() function from the car package. This function also provides the results of a lack-of-fit test for each of these relationships (note when it is the fitted values \(\hat y\) it gets called “Tukey’s test”).
If the assumption is met, you should see a random scatter of \((x,y)\) points with constant mean and variance functions i.e., the vertical spread of the residuals should roughly be the same everywhere.
We can run plot(mymodel) which will cycle through these plots (asking us to press enter each time to move to the next plot), or we can arrange these plots in a matrix via par(mfrow), for example in a 2 x 2 matrix as shown below (make sure to always reset your graphical parameters! If needed, we could also extract specific plots using, for instance: plot(mymodel, which = 3) for the third plot.
Top Left: For the Residuals vs Fitted plot, we want the red line to be horizontal at close to zero across the plot. We don’t want the residuals (the points) to be fanning in/out.
Top Right: For the Normal Q-Q plot, we want the residuals (the points) to follow closely to the diagonal line, indicating that they are relatively normally distributed.2
Bottom Left: For the Scale-Location plot, we want the red line to be horizontal across the plot. These plots allow us to examine the extent to which the variance of the residuals changes across the fitted values. If it is angled, we are likely to see fanning in/out of the points in the residuals vs fitted plot.
Bottom Right: The Residuals vs Leverage plot indicates points that might be of individual interest as they may be unduly influencing the model. There are funnel-shaped lines on this plot (sometimes out of scope of the plotting window). Ideally, we want our residuals inside the funnel - the further the residual is to the right (the more leverage it has), the closer to the 0 we want it to be.
Note, if we have only categorical predictors in our model, many of these will show vertical lines of points. This doesn’t indicate that anything is wrong, and the same principles described above continue to apply
The check_model() function from the performance package is a useful way to check the assumptions of models, as it also returns some useful notes to aid your interpretation. However, it is important to check each assumption individually with plots that are more suitable for a statistics report.
For the linear model with multiple explanatory variables, we need to also think about multicollinearity - this is when two (or more) of the predictors in our regression model are moderately or highly correlated.
We can assess multicollinearity using the variance inflation factor (VIF), which for a given predictor \(x_j\) is calculated as:
\[
VIF_j = \frac{1}{1-R_j^2} \\
\]
Interpretation Guidance
Suggested cut-offs for VIF are varied. Some suggest 10, others 5. Define what you will consider an acceptable value prior to calculating it. You could loosely interpret VIF values \(>5\) as moderate multicollinearity and values \(>10\) as severe multicollinearity.
In R
The vif() function from the car package will provide VIF values for each predictor in your model.
We have seen that some specific individual cases in our data can influence our model more than others. We can identify these as:
Regression outliers: A large residual \(\hat \epsilon_i\) - i.e., a big discrepancy between their predicted y-value and their observed y-value.
Standardised residuals: For residual \(\hat \epsilon_i\), divide by the estimate of the standard deviation of the residuals. In R, the rstandard() function will give you these
Studentised residuals: For residual \(\hat \epsilon_i\), divide by the estimate of the standard deviation of the residuals excluding case \(i\). In R, the rstudent() function will give you these.
High leverage cases: These are cases which have considerable potential to influence the regression model (e.g., cases with an unusual combination of predictor values).
Hat values: are used to assess leverage. In R, The hatvalues() function will retrieve these.
High influence cases: When a case has high leverage and is an outlier, it will have a large influence on the regression model.
Cook’s Distance: combines leverage (hatvalues) with outlying-ness to capture influence: \(D_i = \text{Outlyingness} \times \text{Leverage}\). Cook’s distance refers to the average distance the \(\hat{y}\) values will move if a given case is removed. In R, the cooks.distance() function will provide these values. Alongside Cook’s Distance, we can examine the extent to which model estimates and predictions are affected when an entire case is dropped from the dataset and the model is refitted.
DFFit: the change in the predicted value at the \(i^{th}\) observation with and without the \(i^{th}\) observation is included in the regression.
DFbeta: the change in a specific coefficient with and without the \(i^{th}\) observation is included in the regression. DFbeta represents the difference in the beta coefficients when a case is excluded from the model versus when it’s included. A large DFbeta value would suggest that a case has a substantial impact on the estimated coefficients, and thus a high influence on the model results; a small DFbeta value would suggest that the case has less influence on the estimated coefficients. A commonly used cut-off or threshold to compare \(|DFBETA|\) values (absolute values) against is \(\frac{2}{\sqrt{n}}\) (see Belsley et al., (1980) p. 28 for more info)3.
DFbetas: the change in a specific coefficient divided by the standard error, with and without the \(i^{th}\) observation is included in the regression.
COVRATIO: measures the effect of an observation on the covariance matrix of the parameter estimates. In simpler terms, it captures an observation’s influence on standard errors. Values which are \(>1+\frac{3(k+1)}{n}\) or \(<1-\frac{3(k+1)}{n}\) are considered as having strong influence.
influence.measures(my_model) will give you out a dataframe of the various measures.
summary(influence.measures(my_model)) will provide a nice summary of what R deems to be the influential points.
Next Steps: What to do with Violations of Assumptions / Problematic Case Diagnostic Results
There are lots of different options available, and there is no one right answer. Assuming that we have no issues with model specification (i.e., are not missing variables, have modeled appropriately), then we may want to consider one of the below approaches (note: this is not an exhaustive list!)
The first step is to re-examine your data. It is important to be familiar with your dataset, as you need to know what values are typical, normal, and possible. Could it be the case that you have missed some impossible values (e.g., a negative value of a persons height), values outwith the possible range (e.g., a score of 55 on a survey where scores can only range 10-50), values that don’t make any sense (e.g., an age of 200), or maybe there are even typos / data entry errors (e.g., forgetting to put a decimal point, so having a height of 152m instead of 1.52m)!
If there is a simple error in the data, it could be that you can fix the typo. If that is not possible (maybe you didn’t collect the data, so are unsure of what the value(s) should/could be), you will need to delete the value (i.e., set as an NA), because you know that it is incorrect.
We should aim to never change a legitimate value where possible (and remember that if you have a large dataset, a small number of extreme values will be unlikely to have a strong influence on your results).
If there is an extreme, but legitimate value that you have determined is adversely influencing your model (i.e., by examining the assumptions and diagnostics as outlined above), you may want to consider ways to reduce this influence (e.g., winsorizing - which essentially truncates or caps the identified extreme values to a specified percentile, in turn reducing their influence on the model without completely eliminating the observation(s). For example, you could replace values below the 5th percentile with the 5th percentile value, and values above the 95th percentile with the 95th percentile value).
If after re-examining your data you cannot identify any atypical, non-normal, or impossible values, you may need to select a different approach as outlined below.
This allows us to assess the sensitivity of our results (i.e., parameter estimates, p-values, confidence intervals) to changes in our modelling approach (i.e., the removal of observations).
We can re-fit our model after excluding our identified outliers and potentially influential observations, and compare these results to the original model.
Process of Removing Observations
The current example involves removing all identified outliers and potentially influential observations at the same time. Ideally, and to ensure a more thorough sensitivity analysis, you would remove each of these observations one at a time, assess the effects on the model by comparing to your original, reassessing the remaining pre-identified observations, and repeating the process if necessary.
tab_model(wb_mdl1, wb_mdl2, dv.labels =c("Wellbeing (WEMWBS Scores)", "Wellbeing (WEMWBS Scores)"), pred.labels =c("outdoor_time"="Outdoor Time (hours per week)","social_int"="Social Interactions (number per week)"), title ="Regression Table for Wellbeing Models wb1 and wb2")
Regression Table for Wellbeing Models wb1 and wb2
Wellbeing (WEMWBS Scores)
Wellbeing (WEMWBS Scores)
Predictors
Estimates
CI
p
Estimates
CI
p
(Intercept)
28.62
25.69 – 31.55
<0.001
27.91
25.10 – 30.73
<0.001
Outdoor Time (hours per
week)
0.20
0.10 – 0.30
<0.001
0.19
0.10 – 0.29
<0.001
Social Interactions
(number per week)
0.33
0.16 – 0.51
<0.001
0.40
0.22 – 0.58
<0.001
Observations
200
168
R2 / R2 adjusted
0.126 / 0.118
0.177 / 0.167
We conducted a sensitivity analysis to assess how robust our conclusions were regarding outdoor time and the weekly number of social interactions in the presence of previously identified outliers and potentially influential observations. We re-fit the model, excluding these 28 observations (14% of our original sample), and compared these model results (wb_mdl2) to those of our original model (wb_mdl1).
There was little difference in the estimates from wb_mdl1 and wb_mdl2, and so we can conclude that after conducting a sensitivity analysis, there were no meaningful differences in our results, and hence our conclusions from our original model hold. Specifically:
The direction of all model estimates are the same in wb_mdl1 and wb_mdl2 (i.e., all positive)
There is no difference in statistical significance, and the p-values were of a similar magnitude (i.e., all < .001)
The estimate and confidence intervals for outdoor_time are very similar
There are some quantitative differences in the estimate and confidence intervals for social_int. The estimate differs slightly in magnitude by 0.07), but given that this remains positive and significant, we do not need to be too concerned about this.
The bootstrap method is an alternative non-parametric method of constructing a standard error. Instead of having to rely on calculating the standard error with a formula and potentially applying fancy mathematical corrections, bootstrapping involves mimicking the idea of “repeatedly sampling from the population”. It does so by repeatedly resampling with replacement from our original sample.
What this means is that we don’t have to rely on any assumptions about our model residuals, because we actually generate an actual distribution that we can take as an approximation of our sampling distribution, meaning that we can actually look at where 95% of the distribution falls, without having to rely on any summing of squared deviations.
Note, the bootstrap may provide us with an alternative way of conducting inference, but our model may still be mis-specified. It is also very important to remember that bootstrapping is entirely reliant on utilising our original sample to pretend that it is a population (and mimic sampling from that population). If our original sample is not representative of the population that we’re interested in, bootstrapping doesn’t help us at all.
The method of ordinary least squares regression (OLS: i.e., the type of regression model you have been fitting on the course) assumes that there is constant variance in the errors (homoscedasticity). The method of weighted least squares (WLS) can be used when the ordinary least squares assumption of constant variance in the errors is violated (i.e., you have evidence of heteroscedasticity, like we do in Q3 of this lab).
If we have some specific belief that your non-constant variance is due to differences in the variances of the outcome between various groups, then it might be better to use Weighted Least Squares.
As an example, imagine we are looking at weight of different dog breeds (Figure 12). The weights of chihuahuas are all quite close together (between 2 to 5kg), but the weight of, for example, spaniels is anywhere from 8 to 25kg - a much bigger variance.
Figure 12: The weights of 49 dogs, of 7 breeds
Recall that the default way that lm() deals with categorical predictors such as dog breed, is to compare each one to a reference level. In this case, that reference level is “beagle” (first in the alphabet). Looking at Figure 12 above, which comparison do you feel more confident in?
A: Beagles (14kg) vs Pugs (9.1kg). A difference of 4.9kg.
B: Beagles (14kg) vs Spaniels (19kg). A difference of 5kg.
Hopefully, your intuition is that A looks like a clearer difference than B because there’s less overlap between Beagles and Pugs than between Beagles and Spaniels. Our standard linear model, however, assumes the standard errors are identical for each comparison:
Weighted least squares is a method that allows us to apply weights to each observation, where the size of the weight indicates the precision of the information contained in that observation.
We can, in our dog-breeds example, allocate different weights to each breed. Accordingly, the Chihuahuas are given higher weights (and so Chihuahua comparisons result in a smaller SE), and Spaniels and Retrievers are given lower weights.
library(nlme)load(url("https://uoepsy.github.io/data/dogweight.RData"))dogmod_wls=gls(weight~breed, data =dogdf, weights =varIdent(form =~1|breed))summary(dogmod_wls)
We can also apply weights that change according to continuous predictors (e.g. observations with a smaller value of \(x\) are given more weight than observations with larger values).
A data transformation involves the replacement of a variable (e.g., \(y\)) by a function of that variable in order to change the shape of a distribution or association (e.g., to help reduce skew). We can transform the outcome variable prior to fitting the model, using something such as log(y) or sqrt(y). This will sometimes allow us to estimate a model for which our assumptions are satisfied.
Some of the most common (not an exhaustive list) transformations are:
Log (log(y)): Often used for reducing right skewness. Note, this transformation cannot be applied to zero or negative values (make sure to check your data!)
Square root (sqrt(y)): Also often used for reducing right skewness. This transformation can be applied to zero values (but not negative), and is commonly applied to count data
Figure 13: A model of a transformed outcome variable can sometimes avoid violations of assumptions that arise when modeling the outcome variable directly. Data from https://uoepsy.github.io/data/trouble1.csv
The major downside of this is that we are no longer modelling \(y\), but some transformation \(f(y)\) (\(y\) with some function \(f\) applied to it). Interpretation of the coefficients changes accordingly, such that we are no longer talking in terms of changes in y, but changes in \(f(y)\). When the transformation function used is non-linear (see the Right-Hand of Figure 14) a change in \(f(y)\) is not the same for every \(y\).
Figure 14: The log transformation is non-linear
For certain transformations, we can re-express coefficients to be interpretable with respect to \(y\) itself. For instance, the model using a log transform \(ln(y) = b_0 + b_1(x)\) gives us a coefficient that represents statement A below. We can re-express this by taking the opposite function to logarithm, the exponent, exp(). Similar to how this works in logistic regression, the exponentiated coefficients obtained from exp(coef(model)) are multiplicative, meaning we can say something such as statement B
A: “a 1 unit change in \(x\) is associated with a \(b\) unit change in \(ln(y)\)”.
B: “a 1 unit change in \(x\) is associated with \(e^b\)percent change in \(y\).”
Finding the optimal transformation to use can be difficult, but there are methods out there to help you. One such method is the BoxCox transformation, which can be conducted using BoxCox(variable, lambda="auto"), from the forecast package.4
Generalized Linear Models (GLMs) can appropriately deal with data that do not follow a normal distribution (which is a requirement for traditional linear models). They can accommodate various types of distributions, including the Poisson, binomial, and gamma distributions. This makes them suitable for modelling count data (e.g., number of sunny days Edinburgh has per year - yes, count data can include 0!), binary data (where there are only two possible values e.g., doesn’t wear glasses vs wear glasses, smoker vs non-smoker, i.e., values that are yes/no or 0/1), and other types of non-normal data.
We will explore some GLMs later in the course (Semester 2 Block 4), where we will work with logistic regression models.
Higher order regression terms refer to the inclusion of polynomial terms of degree higher than one in a regression model. In a linear regression model, the association between the dependent variable (\(Y\)) and the independent variable (\(X\)) is assumed to be linear, which means the association can be represented by a straight line. However, in many real-world scenarios, associations between variables are not strictly linear, and including higher order regression terms can help capture more complex relationships. Higher order terms that you could incorporate include quadratic, cubic, or higher degree polynomial terms.
For example, in a quadratic regression model, the relationship between \(Y\) and \(X\) can be represented as:
\[
Y = \beta_0 + \beta_1 \cdot X + \beta_2 \cdot X^2 + \epsilon
\]\[
\begin{align}
& \text{Where:} \\
& Y = \text{Dependent Variable} \\
& X = \text{Independent Variable} \\
\end{align}
\]
As in our models we’ve seen so far, \(\beta_0\), \(\beta_1\), and \(\beta_2\) are the coefficients to be estimated in the above model. What is different from what we’ve seen in DAPR2 is the term \(\beta_2 \cdot X^2\), and this represents the quadratic term. This allows for a curved as opposed to straight line to represent the association between \(Y\) and \(X\), and hence can allow us to capture more complex relationships. For example, we can model the association between height and age:
Figure 15: Two linear models, one with a quadratic term (right)
Please note that these types of models are beyond the scope of the DAPR2 course, but if you want to know more, please do read up on these in your own time.
Removing outliers and potentially influential observations should be a last resort - not all outliers are inherently ‘bad’ - we do expect natural variation in our population(s) of interest. Outliers can be informative about the topic under investigation, and this is why you need to be very careful about excluding outliers due only to their ‘extremeness’. In doing so, you can distort your results by removing variability - i.e., by forcing the data to be more normal and less variable than it actually is, and reduce statistical power by reducing the size of your sample.
If you do decide to remove observations, you will need to document what specific data points you excluded, and provide an explanation as to why these were excluded.
To set specific values to NA in our dataset (and save this updated dataset in a new object named mwdata2), we could use the following code. For the purpose of this demonstration, lets say that we wanted to set any age values of <20 as NA. In the original dataset mwdata, we had 3 individuals aged 18, and 6 aged 19, so we should end up with 9 NA values in mwdata2 column age:
#specify age column in original dataset, where age is < 20, for values to be set to NA and save to new object named mwdata2 to avoid overwriting original datamwdata2<-mwdata|>mutate(age =replace(age, age<20, NA))#check how many NA values we have - there should be 9 (so 9 TRUEs):table(is.na(mwdata2$age))
FALSE TRUE
191 9
If we wanted to remove a full row from the datset, we could use the following code. For the purpose of this demonstration, lets say that we wanted to remove all rows that were highlighted in the above assumption and diagnostic checks as potentially having an adverse influence on our model estimates:
# create new dataset 'mwdata3' without (by specifying -) identified outliers and potentially influential observationsmwdata3<-mwdata[-c(16, 25, 50, 53, 56, 58, 59, 60, 62, 72, 73, 75, 76, 78, 79, 85, 101, 109, 125, 126, 127, 131, 149, 151, 159, 163, 165, 169, 173, 176, 179, 197), ]# check dimensions - should now have 32 rows less than original dataset 200 - 32 = 168dim(mwdata3)
[1] 168 7
Bootstrap
The bootstrap is a general approach to assessing whether the sample results are statistically significant or not, and allows us to draw inferences to the population from a regression model. This method is assumption-free and does not rely on conditions such as normality of the residuals.
It is based on sampling repeatedly with replacement (to avoid always getting the original sample exactly) from the data at hand, and then computing the regression coefficients from each re-sample. We will equivalently use the word “bootstrap sample” or “resample” (for sample with replacement).
Overview
The basic principle is:
The population is to the original sample
as
the original sample is to the bootstrap samples.
Because we only have one sample of size \(n\), and we do not have access to the data for the entire population, we consider our original sample as our best approximation to the population.
To be more precise, we assume that the population is made up of many, many copies of our original sample. Then, we take multiple samples each of size \(n\) from this assumed population. This is equivalent to sampling with replacement from the original sample.
Terminology
A parameter is a numerical summary for the population, e.g. the population slope \(\beta_1\).
A statistic is a numerical summary calculated from the sample data, e.g. the estimated slope in the sample \(\widehat \beta_1\). We use the sample statistic as a best guess, or estimate, for the unknown population parameter.
A bootstrap sample is chosen with replacement from an existing sample, using the same sample size.
A bootstrap statistic is a statistic computed for each bootstrap sample.
A bootstrap distribution collects bootstrap statistics for many bootstrap samples.
In R
Follow these steps:
1: Load the car package.
2: Use the Boot() function (do not forget the uppercase B!) which takes as arguments:
the fitted model
f, saying which bootstrap statistics to compute on each bootstrap sample. By default f = coef, returning the regression coefficients.
R, saying how many bootstrap samples to compute. By default R = 999 but this could be any number. To experiment we recommend 1000, when you want to produce results for journals, it is typical to go with 10,000 or more.
ncores, saying if to perform the calculations in parallel (and more efficiently). However, this will depend on your PC, and you need to find how many cores you have by running parallel::detectCores() on your PC. By default the function uses ncores = 1.
3: Run the code. However, please remember that the Boot() function does not want a model which was fitted using data with NAs. To remove, for example, you could use na.omit.
4: Look at the summary() of the bootstrap results. When doing so the output will show, for each regression coefficient, the value in the original sample in the column original, and in the bootSE column, the estimate of the variability of the coefficient from bootstrap sample to bootstrap sample. The bootSE provides us the bootstrap standard error, or bootstrap SE in short. We can use this to answer the key question of how accurate our estimate is.
5: Compute confidence intervals via the Confint() function. Use your preferred confidence level (usually, and by default, 95%) by specifying level =. If you select 95% confidence intervals, by also specifying the type = "perc" argument, R will return the values that comprise 95% of all values in between them, i.e. the value with 2.5% of observations below it and the value with 2.5% of observations above it and 97.5% of observations below it.
6: Provide interpretation in the context of your research question and report results in APA format. (Note: the actual estimates are those from our original model, it is just the bounds of the interval that bootstrapping is providing us with).
In R
#specify modelrecall_mdl<-lm(recall_accuracy~recall_confidence+age, data =recalldata)#step 1: load car packagelibrary(car)#step 2/3: bootstrap model (asking to resample 1000 times, i.e., getting a distribution of 1000 values for the coefficients)bootmymodel<-Boot(recall_mdl, R =1000)#step 4: check summarysummary(bootmymodel)
Number of bootstrap replications R = 1000
original bootBias bootSE bootMed
(Intercept) 36.15959 -3.2711632 16.47382 34.86127
recall_confidence 0.89573 0.0522770 0.25721 0.92141
age -0.33916 0.0027399 0.11400 -0.32973
#step 5: confidence intervalsConfint(bootmymodel, level =0.95, type ="perc")
You can visualise the uncertainty in the estimates by plotting histograms using the built-in function from the car package, which simply takes the bootstrap results bootmymodel. The bootstrap distribution should appear to be normal, and if non-normal, results are likely untrustworthy (note the distribution can be somewhat ambiguous with a small number of resamples).
By embedding LaTeX into RMarkdown, you can accurately and precisely format mathematical expressions, ensuring that they are not only technically correct but also visually appealing and easy to interpret.
APA format is a writing/presentation style that is often used in psychology to ensure consistency in communication. APA formatting applies to all aspects of writing - from formatting of papers (including tables and figures), citation of sources, and reference lists. This means that it also applies to how you present results in your Psychology courses, including DAPR2.
APA Formatting Guides
All results should be presented following APA guidelines.
Make sure to familiarise yourself with the above guides, and practice presenting your results following these rules.
Tables
We want to ensure that we are presenting results in a well formatted table. To do so, there are lots of different packages available (see Lesson 4 of the RMD bootcamp).
One of the most convenient ways to present results from regression models is to use the tab_model() function from sjPlot.
Creating tables via tab_model
Within tab_model(), there are lots of different ways that you can customise your table. The most common arguments that you should use are dv.labels, pred.labels, and title.
You can rename your DV and IV labels by specifying dv.labels and pred.labels. To do so, specify your variable name on the left, and what you would like this to be named in the table on the right. For title, you can simply specify in ““’s what you want your title to be e.g., title = "This is my title".
Here’s an example if I had fitted a model with the following information:
Model name = mdl_test
Model DV = cognitive_score
Model IVs = SES and age
mdl_test<-lm(cognitive_score~SES+age, data =data_name)
I want to change the names of SES and age to be socio-economic status and age - in years respectively. What we need to pay attention to here is the ordering of the IVs - the ordering in our lm() must match that in tab_model(). I also want to name my table Regression Table for Cognitive Scores Model. Here is how we would do this in R:
library(sjPlot)tab_model(mdl_test, pred.labels =c('Intercept', 'socio-economic status', 'age - in years'), title ="Regression Table for Cognitive Scores Model")
Cross-referencing is a very helpful way to direct your reader through your document, and the good news is that this can be done automatically in RMarkdown.
Cross Referencing
There are three key components to allow you to successfully cross-reference within your RMarkdown document:
A bookdown output format
A caption to your figure or table
A named/labeled code chunk
Once you have the above, you will be able to cross-reference using the syntax @ref(type:label), where label is the chunk name/label, and type is the environment being referenced (e.g. tab for table, fig for figure, etc.).
Yes, the error term is gone. This is because the line of best-fit gives you the prediction of the average recall accuracy for a given age, and not the individual recall accuracy of an individual person, which will almost surely be different from the prediction of the line.↩︎
QQplots plot the values against the associated percentiles of the normal distribution. So if we had ten values, it would order them lowest to highest, then plot them on the y against the 10th, 20th, 30th.. and so on percentiles of the standard normal distribution (mean 0, SD 1)↩︎
Belsley, D. A., Kuh, E., & Welsch, R. E. (2005). Regression diagnostics: Identifying influential data and sources of collinearity. John Wiley & Sons. DOI: 10.1002/0471725153↩︎
This method finds an appropriate value for \(\lambda\) such that the transformation \((sign(x) |x|^{\lambda}-1)/\lambda\) results in a close to normal distribution.↩︎